Advanced Stereoselective Synthesis of Cis-Nucleosides for Commercial Antiviral Production
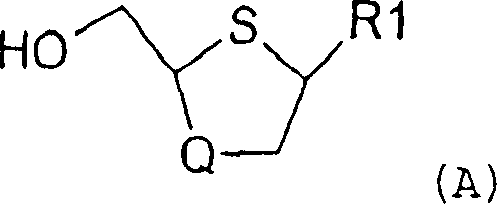
The pharmaceutical industry continuously seeks robust and efficient synthetic routes for critical antiviral intermediates, particularly those targeting HIV and Hepatitis B viruses. Patent CN1297553C discloses a groundbreaking stereoselective method for the preparation of nucleosides and nucleoside analogues of Formula (A), which have demonstrated potent inhibitory activity against HIV-1 replication and Hepatitis B virus infection. This technology represents a significant leap forward from traditional multi-step syntheses by introducing a streamlined catalytic coupling strategy that directly links a sulfoxide precursor with a pyrimidine base. The core innovation lies in the ability to predominantly generate the biologically active cis-isomer through precise control of reaction parameters and catalytic systems. By leveraging specific Group IB or IIB elements alongside Lewis acids, this process achieves high stereochemical fidelity without the need for cumbersome protecting group manipulations typically seen in earlier methodologies. For procurement and R&D teams, understanding this patented pathway is essential for securing a reliable nucleoside intermediate supplier capable of delivering high-purity materials for next-generation antiviral therapies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-substituted, 4-substituted 1,3-oxathiolane nucleosides has been plagued by inefficiencies inherent in multi-step linear sequences. Prior art methods, such as those described in PCT publications WO92/08717 and WO95/29176, often require extensive protection and deprotection cycles to manage the reactivity of hydroxyl and amino groups. These traditional routes frequently suffer from reduced overall yields due to material loss at each isolation stage and the generation of significant chemical waste. Furthermore, achieving high stereoselectivity for the cis-isomer often necessitates complex chromatographic separations or resolution steps, which are costly and difficult to scale for commercial production. The reliance on expensive starting materials and stoichiometric reagents further exacerbates the cost burden, making cost reduction in antiviral drug manufacturing a persistent challenge for supply chain managers. Consequently, there is a critical need for a more direct and atom-economical approach that minimizes operational complexity while maximizing the output of the therapeutically relevant isomer.
The Novel Approach
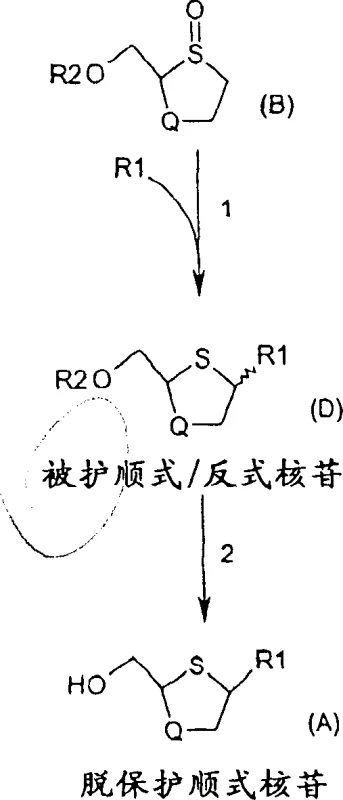
The methodology outlined in Patent CN1297553C offers a transformative solution by condensing the synthesis into a highly efficient two-step process centered around a catalytic coupling reaction. Instead of laborious sequential modifications, this novel approach involves the direct coupling of a sulfoxide molecule of Formula (B) with a pyrimidine base (R1) in a single pot. The reaction is facilitated by a sophisticated catalytic system comprising a tertiary amine, a Lewis acid, and catalytic amounts of Group IB or IIB elements. This strategic combination allows for the formation of the intermediate Formula (D) with a cis-to-trans ratio greater than 2:1, significantly enriching the desired stereoisomer right from the coupling stage. The subsequent deprotection step is ingeniously designed to not only remove protecting groups but also to facilitate the selective crystallization of the cis-nucleoside, thereby enhancing purity without additional purification columns. This streamlined workflow drastically simplifies the commercial scale-up of complex pharmaceutical intermediates, offering a clear pathway to higher throughput and lower production costs.
Mechanistic Insights into Cu/Zn-Catalyzed Stereoselective Coupling
The heart of this technological advancement lies in the mechanistic interplay between the transition metal catalyst and the Lewis acid activator. The process utilizes elements from Group IB or IIB of the periodic table, specifically highlighting the efficacy of copper (Cu+, Cu2+) and zinc (Zn2+) species such as CuCl, CuCl2, and ZnBr2. These metals act as catalysts to promote the nucleophilic attack of the silylated pyrimidine base onto the activated sulfoxide electrophile. The presence of a Lewis acid, such as trimethylsilyl triflate (TMSOTf) or trimethylsilyl iodide (TMSI), is crucial for activating the sulfoxide oxygen, rendering the adjacent carbon more susceptible to substitution. Crucially, the stereochemical outcome is not left to chance but is rigorously controlled by thermodynamic and kinetic factors, primarily the reaction temperature. The patent data explicitly states an inverse relationship between the coupling temperature and the cis-to-trans isomer ratio, meaning that conducting the reaction at lower temperatures, ranging from approximately 0°C to -50°C, significantly favors the formation of the cis-configuration. This temperature-dependent stereoselectivity allows manufacturers to tune the process to maximize the yield of the active pharmaceutical ingredient precursor.
Furthermore, the impurity profile is managed effectively through the choice of solvent and the specific conditions of the deprotection step. The intermediate Formula (D), which contains a mixture of cis and trans isomers, undergoes deprotection in a solvent system chosen to favor the crystallization of the cis-product. Solvents such as methanol, ethanol, or mixtures with water or toluene are employed to precipitate the desired cis-nucleoside while leaving impurities or the trans-isomer in the mother liquor. This crystallization-driven purification is a powerful tool for ensuring high-purity cis-nucleosides without the need for preparative HPLC, which is often a bottleneck in large-scale manufacturing. The ability to use enantiomerically pure starting sulfoxides, as shown in specific embodiments, further ensures that the final product maintains the necessary optical purity required for clinical applications. This level of control over both diastereoselectivity and enantioselectivity underscores the robustness of the method for producing clinical-grade materials.
How to Synthesize Cis-Nucleoside Efficiently
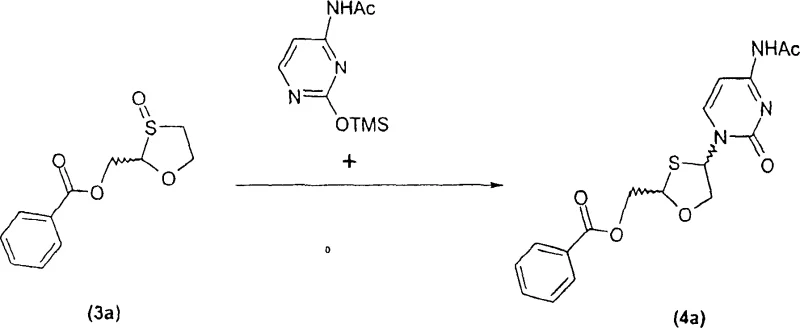
Implementing this synthesis requires careful attention to the order of addition and temperature control during the coupling phase to ensure optimal stereoselectivity. The detailed standardized synthesis steps involve dissolving the protected sulfoxide in a chlorinated solvent like dichloromethane, cooling the mixture, and sequentially adding the amine and silylating agent before introducing the metal catalyst and base. Following the coupling, the reaction mixture is worked up and subjected to basic conditions to remove protecting groups, inducing crystallization of the final product. For a complete breakdown of the specific reagent quantities, timing, and workup procedures required to replicate this high-yield process, please refer to the structured guide below.
- Couple a protected sulfoxide intermediate (Formula B) with a pyrimidine base (R1) in the presence of a catalytic amount of Group IB or IIB elements (e.g., Cu, Zn), a tertiary amine, and a Lewis acid to form intermediate Formula D.
- Perform a deprotection step on intermediate Formula D using a suitable deprotecting agent and solvent system to selectively crystallize and isolate the target cis-nucleoside (Formula A).
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthetic route offers substantial strategic advantages beyond mere technical elegance. The reduction in the number of synthetic steps directly correlates to a significant decrease in raw material consumption, solvent usage, and waste disposal costs, driving down the overall cost of goods sold. By eliminating the need for expensive chiral resolving agents or complex chromatographic separations in the final stages, the process becomes inherently more cost-effective and easier to validate for regulatory compliance. The reliance on commodity chemicals such as copper salts, common amines, and standard chlorinated solvents ensures that the supply chain remains resilient against fluctuations in the availability of exotic reagents. This stability is crucial for maintaining continuous production schedules and meeting the demanding delivery timelines of global pharmaceutical clients.
- Cost Reduction in Manufacturing: The streamlined two-step process eliminates multiple protection and deprotection cycles found in conventional methods, which drastically reduces solvent volumes and labor hours associated with intermediate isolations. By utilizing catalytic amounts of inexpensive transition metals like copper instead of stoichiometric quantities of precious metals or reagents, the direct material costs are significantly lowered. The ability to purify the final product through crystallization rather than chromatography further reduces operational expenses related to silica gel and solvent recovery. These cumulative efficiencies result in a much leaner manufacturing process that offers substantial cost savings without compromising on the quality or purity of the nucleoside intermediate.
- Enhanced Supply Chain Reliability: The reagents required for this synthesis, including tertiary amines, Lewis acids, and copper salts, are widely available from multiple global chemical suppliers, reducing the risk of single-source bottlenecks. The robustness of the reaction conditions allows for flexibility in sourcing, as the process tolerates standard industrial grade solvents and reagents without requiring ultra-high purity inputs for the initial steps. This flexibility ensures that production can continue uninterrupted even if specific vendor supplies are temporarily constrained, providing a secure and reliable nucleoside intermediate supplier partnership. Additionally, the simplified workflow reduces the lead time for high-purity nucleoside intermediates, allowing for faster response to market demand surges.
- Scalability and Environmental Compliance: The process is designed with commercial scale-up in mind, utilizing reaction conditions that are easily transferable from laboratory to pilot and production scales without exothermic runaway risks. The crystallization-based purification minimizes the generation of liquid waste streams associated with column chromatography, aligning with modern green chemistry principles and environmental regulations. Reduced solvent intensity and the elimination of heavy metal waste from stoichiometric reactions simplify wastewater treatment and disposal protocols. This environmental efficiency not only lowers compliance costs but also enhances the sustainability profile of the manufactured antiviral ingredients, appealing to eco-conscious pharmaceutical partners.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation and benefits of this stereoselective synthesis technology. These insights are derived directly from the experimental data and claims within the patent documentation to provide accurate guidance for potential partners. Understanding these details helps in evaluating the feasibility of integrating this route into existing manufacturing portfolios for antiviral drug production.
Q: How is stereoselectivity controlled in this nucleoside synthesis?
A: Stereoselectivity is primarily controlled by the reaction temperature during the coupling step. The patent data indicates an inverse relationship between temperature and the cis-to-trans ratio, with lower temperatures (e.g., -15°C to -50°C) favoring the formation of the desired cis-isomer.
Q: What catalysts are utilized in this novel coupling method?
A: The process employs catalytic amounts of Group IB or Group IIB elements, specifically citing copper (Cu+, Cu2+) and zinc (Zn2+) species such as CuCl, CuCl2, and ZnBr2, often in combination with silyl-based Lewis acids.
Q: Does this method support large-scale commercial manufacturing?
A: Yes, the method is designed for scalability by minimizing protection-deprotection sequences and utilizing a crystallization-driven purification in the final deprotection step, which simplifies downstream processing and enhances overall yield.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Cis-Nucleoside Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthetic routes in the competitive landscape of antiviral drug development. Our team of expert chemists has extensively evaluated the methodology described in Patent CN1297553C and possesses the technical capability to adapt and optimize this catalytic coupling strategy for your specific project needs. We have extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from clinical trials to market launch is seamless and supported by a robust supply chain. Our state-of-the-art facilities are equipped with rigorous QC labs and stringent purity specifications to guarantee that every batch of cis-nucleoside intermediate meets the highest international regulatory standards.
We invite you to collaborate with us to leverage this advanced technology for your next-generation antiviral programs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements, demonstrating how this streamlined process can improve your margins. Please contact us today to request specific COA data and route feasibility assessments, and let us help you secure a competitive advantage in the global pharmaceutical market through superior chemical manufacturing solutions.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →