Scalable Synthesis of Spiro-Octane Carboxylic Acid Intermediates for Commercial API Production
Introduction to Patent CN111574524A and Strategic Process Innovation
The pharmaceutical industry constantly seeks robust synthetic routes for complex heterocyclic scaffolds, particularly spiro-cyclic systems which are increasingly prevalent in modern drug discovery portfolios. Patent CN111574524A addresses a critical gap in the supply chain by disclosing a novel, four-step preparation method for 2-(tert-butoxycarbonyl)-7-oxyidene-2,6-diazaspiro[3.4]octane-5-carboxylic acid (CAS: 1357351-94-0). Historically, the synthesis of such densely functionalized spiro-azetidine derivatives has been plagued by low yields, difficult purification, and a reliance on scarce starting materials, creating significant bottlenecks for reliable pharmaceutical intermediate supplier networks. This patent introduces a streamlined approach that leverages readily available precursors and standard unit operations to achieve a scalable pathway. By utilizing a sequence of Michael addition, catalytic hydrogenation, base-mediated cyclization, and hydrolysis, the technology offers a viable solution for cost reduction in API manufacturing where previous methods failed to meet industrial throughput requirements.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the innovations detailed in this patent, the synthesis of 2,6-diazaspiro[3.4]octane derivatives was largely confined to laboratory-scale explorations with limited practical utility for commercial production. Existing literature often describes routes that involve harsh reaction conditions, unstable intermediates, or multi-step sequences with poor atom economy, leading to excessive waste generation and high operational expenditures. The scarcity of reported methods indicates that traditional approaches struggle with the steric hindrance inherent in forming the spiro-junction between the four-membered azetidine ring and the five-membered lactam ring. Furthermore, conventional pathways frequently require expensive transition metal catalysts or cryogenic conditions that are energy-intensive and difficult to maintain in large-scale reactors. These technical barriers result in inconsistent batch quality and prolonged lead times, making it challenging for procurement teams to secure a steady supply of high-purity pharmaceutical intermediates needed for clinical and commercial programs.
The Novel Approach
The methodology presented in CN111574524A overcomes these historical challenges through a cleverly designed cascade that builds complexity efficiently from simple precursors. The process initiates with a condensation reaction using ethyl nitroacetate and DBU in acetonitrile, establishing the carbon framework necessary for subsequent ring closure. A key differentiator is the use of Raney nickel for hydrogenation, a cost-effective and widely available catalyst that replaces more precious metals while effectively reducing the nitro functionality to the requisite amine for cyclization. The subsequent intramolecular cyclization driven by sodium ethoxide in ethanol proceeds under moderate thermal conditions (80°C), demonstrating excellent tolerance for the sensitive Boc-protecting group. This strategic selection of reagents and conditions ensures that the commercial scale-up of complex pharmaceutical intermediates is feasible without requiring specialized high-pressure equipment beyond standard hydrogenation reactors, thereby significantly de-risking the manufacturing process.
Mechanistic Insights into Base-Mediated Cyclization and Hydrogenation
The core chemical transformation in this synthesis relies on a precise sequence of nucleophilic attacks and reductions that construct the spiro-center with high fidelity. In the initial step, the base DBU facilitates the deprotonation of ethyl nitroacetate, generating a nucleophile that attacks the electrophilic exocyclic double bond of the azetidine precursor via a Michael-type addition. This forms a linear intermediate containing both nitro and ester functionalities, setting the stage for the critical ring-closing event. Following the reduction of the nitro group to an amine using hydrogen gas and Raney nickel, the molecule possesses the necessary nucleophilic nitrogen to attack the adjacent ester carbonyl. The addition of sodium ethoxide serves a dual purpose: it neutralizes any acidic byproducts and activates the amine for nucleophilic acyl substitution, driving the formation of the five-membered lactam ring fused to the azetidine core. This mechanistic pathway is highly favorable entropically as it forms a stable spiro-system, minimizing the formation of oligomeric byproducts that often plague intermolecular reactions.
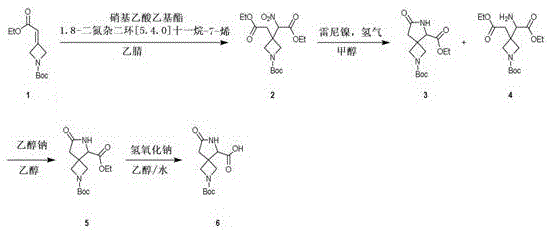
Impurity control is rigorously managed throughout the synthetic sequence to ensure the final product meets stringent purity specifications required for GMP manufacturing. During the hydrogenation step, the use of methanol as a solvent ensures good solubility of the intermediate while allowing for efficient filtration of the Raney nickel catalyst, preventing metal contamination in downstream steps. The cyclization step generates a crude mixture that is purified via column chromatography to isolate the desired spiro-lactam ester (Compound 5) from potential regioisomers or unreacted starting materials. Finally, the hydrolysis step employs a biphasic system of ethanol and water with sodium hydroxide, which selectively cleaves the ethyl ester while preserving the tert-butoxycarbonyl (Boc) protecting group due to the specific pH and temperature controls (80°C). The final acidification to pH 5 precipitates the product, effectively separating it from inorganic salts and water-soluble impurities, resulting in a high-purity solid suitable for further coupling reactions in API synthesis.
How to Synthesize 2-(tert-Butoxycarbonyl)-7-oxyidene-2,6-diazaspiro[3.4]octane-5-carboxylic acid Efficiently
Executing this synthesis requires strict adherence to the thermal and stoichiometric parameters defined in the patent to maximize yield and minimize side reactions. The process begins with the preparation of the nitro-intermediate in acetonitrile, followed by a high-pressure hydrogenation step that demands careful monitoring of hydrogen uptake to ensure complete reduction. Operators must then proceed immediately to the cyclization step using freshly prepared sodium ethoxide to avoid moisture degradation of the base. The final hydrolysis is a straightforward saponification but requires precise pH adjustment during workup to ensure maximum recovery of the free acid. For detailed operational parameters, safety guidelines, and specific stoichiometric ratios, please refer to the standardized synthesis protocol outlined below.
- React tert-butyl 3-(2-ethoxy-2-oxoethylidene)azetidine-1-carboxylate with ethyl nitroacetate and DBU in acetonitrile at 80°C for 12 hours to form the nitro-intermediate.
- Perform catalytic hydrogenation using Raney nickel in methanol at 80°C under 1.2 MPa hydrogen pressure for 6 hours to reduce the nitro group.
- Induce cyclization by treating the reduced intermediate mixture with sodium ethoxide in ethanol at 80°C for 3 hours to form the spiro-lactam ring.
- Hydrolyze the ethyl ester using sodium hydroxide in an ethanol-water mixture at 80°C for 6 hours, followed by acidification to isolate the final carboxylic acid.
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain perspective, this patented route offers substantial advantages by utilizing commodity chemicals and avoiding supply-constrained reagents. The starting material, tert-butyl 3-(2-ethoxy-2-oxoethylidene)azetidine-1-carboxylate, is derived from common building blocks, ensuring that raw material sourcing remains stable even during market fluctuations. The reliance on Raney nickel instead of palladium or platinum catalysts represents a significant strategic benefit, as it eliminates the need for complex metal scavenging processes and reduces the overall cost of goods sold (COGS). Furthermore, the solvents employed—acetonitrile, methanol, and ethanol—are all widely available industrial grade solvents with established recycling protocols, facilitating environmental compliance and waste management. This alignment with green chemistry principles enhances the sustainability profile of the manufacturing process, a key metric for modern procurement evaluations.
- Cost Reduction in Manufacturing: The elimination of expensive noble metal catalysts and the use of ambient pressure conditions for most steps drastically lower capital and operational expenditures. By avoiding cryogenic temperatures and utilizing standard heating mantles or jacketed reactors at 80°C, the energy consumption is optimized, leading to substantial cost savings in utility bills. Additionally, the high selectivity of the cyclization step reduces the burden on purification units, minimizing solvent usage and chromatography resin costs associated with removing complex impurity profiles.
- Enhanced Supply Chain Reliability: The robustness of the four-step sequence ensures consistent batch-to-batch reproducibility, which is critical for maintaining continuous API production schedules. Since the reagents are non-proprietary and commercially available from multiple global vendors, the risk of single-source supply disruption is mitigated. The process design allows for flexible batch sizing, enabling manufacturers to respond quickly to changes in demand without lengthy re-validation periods, thus reducing lead time for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: The reaction conditions are inherently safe and scalable, with no exothermic runaways identified in the patent data, allowing for direct translation from pilot plant to multi-ton production. The aqueous workup in the final step simplifies waste stream treatment, as the effluent primarily contains benign salts and alcohols that are easily treated in standard wastewater facilities. This ease of scale-up ensures that the commercial supply of this critical spiro-intermediate can meet the growing demands of the pharmaceutical sector without compromising on environmental standards.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthetic route. Understanding these details is crucial for process chemists and engineering teams evaluating the feasibility of adopting this technology for their specific production lines. The answers are derived directly from the experimental data and beneficial effects described in the patent documentation.
Q: What are the critical reaction conditions for the cyclization step?
A: The cyclization step requires sodium ethoxide in ethanol at 80°C for 3 hours. Precise temperature control is essential to ensure complete ring closure without degrading the sensitive spiro-framework.
Q: How is the purity of the final spiro-acid ensured?
A: Purity is managed through a combination of column chromatography after the initial steps and selective extraction/crystallization during the final hydrolysis and acidification stages, ensuring removal of residual esters and salts.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the patent explicitly designs the route for industrial scalability, utilizing common solvents like acetonitrile, methanol, and ethanol, and avoiding exotic catalysts in favor of standard Raney nickel.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-(tert-Butoxycarbonyl)-7-oxyidene-2,6-diazaspiro[3.4]octane-5-carboxylic acid Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of reliable access to complex spiro-cyclic intermediates for the development of next-generation therapeutics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. We are committed to delivering products that meet stringent purity specifications through our rigorous QC labs, which utilize advanced analytical techniques to verify identity and assay. By leveraging the efficient synthesis route described in CN111574524A, we can offer competitive pricing and reliable delivery schedules for this high-value building block.
We invite potential partners to engage with our technical procurement team to discuss how we can support your specific project requirements. Whether you need a Customized Cost-Saving Analysis for your current supply chain or require specific COA data and route feasibility assessments for new analogs, our experts are ready to provide tailored solutions. Contact us today to secure a stable supply of this critical intermediate and accelerate your drug development timeline.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →