Advanced Synthesis of 2-Fluoro-3-Methyl-4-Trifluoromethyl Benzylamine Hydrochloride for Commercial Scale-Up
The pharmaceutical industry constantly demands novel intermediates that combine structural complexity with manufacturing feasibility. Patent CN112194558B, published in May 2022, introduces a significant breakthrough in the synthesis of 2-fluoro-3-methyl-4-(trifluoromethyl)benzylamine hydrochloride, a critical building block for next-generation therapeutic agents. This patent outlines a concise, three-step synthetic pathway that overcomes historical challenges associated with regioselective functionalization on highly substituted aromatic rings. By leveraging directed ortho-metalation strategies followed by modern palladium-catalyzed cross-coupling, the disclosed method achieves exceptional control over impurity profiles while maintaining high overall yields. For R&D directors and procurement specialists alike, this technology represents a shift from laborious, low-yielding legacy routes to a streamlined process optimized for both purity and cost-efficiency. The ability to access this specific fluorinated scaffold reliably is paramount for companies developing kinase inhibitors or metabolic modulators where the trifluoromethyl group plays a crucial role in binding affinity and metabolic stability.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of polysubstituted benzylamines bearing both fluoro and trifluoromethyl groups has been plagued by issues of regiocontrol and harsh reaction conditions. Traditional electrophilic aromatic substitution often fails to distinguish between the activated positions on the ring, leading to complex mixtures of isomers that are notoriously difficult to separate, thereby driving up purification costs and reducing overall throughput. Furthermore, earlier methods frequently relied on stoichiometric amounts of hazardous reagents or required extreme temperatures that posed significant safety risks during scale-up. The lack of a standardized, robust protocol meant that supply chains were vulnerable to batch-to-batch variability, with impurities often carrying through to the final API, necessitating expensive remedial processing. In many documented cases, the installation of the methyl group adjacent to a fluorine atom without displacing the halogen or affecting the sensitive trifluoromethyl moiety was a bottleneck that limited the commercial viability of potential drug candidates relying on this scaffold.
The Novel Approach
The methodology described in CN112194558B elegantly circumvents these obstacles through a rational design that prioritizes chemoselectivity and operational simplicity. The innovation lies in the sequential application of a directed lithiation event followed by a mild palladium-catalyzed amination. By utilizing a specific diamine ligand in the first step, the process directs the lithiation exclusively to the desired position, allowing for the clean introduction of the methyl group via methyl iodide quenching. This is immediately followed by a Buchwald-Hartwig coupling that installs the nitrogen functionality under relatively mild thermal conditions. This approach not only simplifies the purification workflow but also significantly enhances the safety profile of the manufacturing process. The result is a route that transforms readily available starting materials into the target hydrochloride salt with minimal waste generation, addressing the core pain points of both process chemistry teams and supply chain managers who require predictable, high-quality outputs.
Mechanistic Insights into Directed Ortho-Lithiation and Pd-Catalyzed Amination
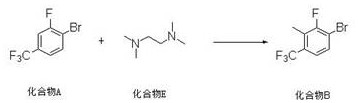
The first stage of this synthesis is a masterclass in organometallic control, utilizing the electronic properties of the fluorine substituent to guide reactivity. In the presence of a chelating diamine ligand, n-butyllithium forms a superbasic complex that selectively deprotonates the aromatic ring at the position ortho to the fluorine atom. This regioselectivity is driven by the coordination of the lithium cation to the fluorine lone pairs, effectively blocking other positions and activating the C3-H bond for abstraction. Once the aryl lithium species is generated at cryogenic temperatures ranging from -80°C to -75°C, it acts as a potent nucleophile. The subsequent addition of methyl iodide results in a rapid SN2-type displacement, installing the methyl group with precision. This step is critical because it establishes the substitution pattern early in the sequence, preventing the formation of difficult-to-remove regioisomers that would otherwise compromise the purity of the final pharmaceutical intermediate.
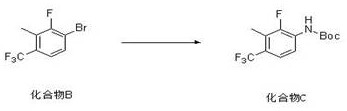
Following the construction of the carbon skeleton, the second step employs a sophisticated palladium catalytic cycle to forge the carbon-nitrogen bond. The reaction utilizes tris(dibenzylideneacetone)dipalladium (Pd2(dba)3) paired with the bulky, electron-rich ligand Xantphos (4,5-bis(diphenylphosphino)-9,9-dimethylxanthene). This specific ligand architecture is essential for stabilizing the active palladium(0) species and facilitating the oxidative addition into the carbon-bromine bond of the intermediate. The wide bite angle of Xantphos promotes the reductive elimination step, which is often the rate-determining step in aminating sterically hindered substrates. Potassium carbonate serves as the base to deprotonate the tert-butyl carbamate, generating the nucleophilic carbamate anion that attacks the palladium center. This catalytic system is remarkably tolerant of the trifluoromethyl group and operates efficiently in dioxane, providing a clean conversion to the protected amine without requiring excessive catalyst loading or prolonged reaction times.
How to Synthesize 2-Fluoro-3-Methyl-4-(Trifluoromethyl)Benzylamine Efficiently
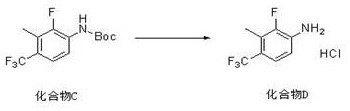
The execution of this synthesis requires careful attention to temperature control and atmosphere management, particularly during the lithiation phase. The process begins by charging a reactor with the bromo-fluoro starting material and the diamine ligand in tetrahydrofuran, followed by cooling to cryogenic conditions before the slow addition of n-butyllithium. After the lithiation is complete, methyl iodide is introduced, and the mixture is allowed to warm gradually to ensure complete conversion. The resulting intermediate is then subjected to the palladium-catalyzed coupling with tert-butyl carbamate under an inert nitrogen atmosphere to prevent catalyst oxidation. Finally, the protecting group is removed using ethanolic hydrochloric acid, precipitating the product as a stable hydrochloride salt. For detailed operational parameters, stoichiometry, and specific workup instructions, please refer to the standardized synthesis guide below.
- Perform directed ortho-lithiation of 1-bromo-2-fluoro-4-(trifluoromethyl)benzene using n-butyllithium and a diamine ligand at cryogenic temperatures, followed by quenching with methyl iodide to install the methyl group.
- Execute a Buchwald-Hartwig cross-coupling reaction between the brominated intermediate and tert-butyl carbamate using a Pd2(dba)3/Xantphos catalyst system in dioxane.
- Deprotect the Boc-group and form the hydrochloride salt by treating the protected amine with ethanolic hydrochloric acid under controlled heating conditions.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented route offers substantial advantages that directly impact the bottom line and supply security for pharmaceutical manufacturers. The reliance on commodity chemicals such as methyl iodide, n-butyllithium, and potassium carbonate means that the raw material supply chain is robust and resistant to market fluctuations. Unlike processes that depend on exotic or custom-synthesized reagents, this method leverages a global supply network of bulk chemicals, ensuring that production schedules are not disrupted by vendor shortages. Furthermore, the high efficiency of the catalytic steps reduces the consumption of expensive palladium resources, while the straightforward isolation procedures minimize solvent usage and waste disposal costs. This alignment with green chemistry principles not only lowers operational expenditures but also simplifies regulatory compliance regarding environmental emissions and hazardous waste handling.
- Cost Reduction in Manufacturing: The elimination of complex purification steps, such as preparative HPLC or multiple recrystallizations, drastically reduces the cost of goods sold. By achieving high purity directly from the reaction workup through simple extraction and filtration, the process minimizes material loss and labor hours. The use of a highly active catalyst system allows for lower catalyst loadings while maintaining excellent conversion rates, which significantly lowers the cost associated with precious metal recovery and refining. Additionally, the high overall yield across the three steps means that less starting material is required to produce a kilogram of the final API intermediate, compounding the savings throughout the production volume.
- Enhanced Supply Chain Reliability: The modular nature of this synthesis allows for flexible manufacturing strategies, where intermediates can be stocked at various stages to buffer against demand spikes. Since the starting materials are widely produced by multiple chemical suppliers globally, the risk of single-source dependency is virtually eliminated. The robustness of the reaction conditions, particularly the tolerance of the palladium catalyst to functional groups, ensures consistent batch quality, reducing the incidence of out-of-specification results that can delay shipments. This reliability is crucial for maintaining just-in-time inventory levels and meeting the rigorous delivery timelines expected by downstream pharmaceutical partners.
- Scalability and Environmental Compliance: The process is designed with scale-up in mind, utilizing solvents like tetrahydrofuran, dioxane, and ethanol which are well-understood in large-scale chemical engineering contexts. The exothermic nature of the lithiation is managed through controlled dosing at low temperatures, a standard unit operation in modern multipurpose plants. Moreover, the final salt formation step produces a solid product that is easy to handle, package, and transport, reducing logistical complexities. The reduction in heavy metal waste and the use of recyclable solvents contribute to a lower environmental footprint, aligning with the increasing corporate sustainability goals of major pharmaceutical buyers and facilitating smoother environmental impact assessments.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthetic route. These insights are derived directly from the experimental data and technical specifications outlined in the patent documentation, providing clarity on process capabilities and product quality. Understanding these details is essential for technical teams evaluating the feasibility of adopting this method for their specific supply chain requirements.
Q: What is the key advantage of the lithiation strategy in this synthesis?
A: The use of a diamine ligand facilitates highly regioselective ortho-lithiation directed by the fluorine atom, ensuring the methyl group is installed precisely at the 3-position without affecting the bromine or trifluoromethyl groups.
Q: How does this method improve supply chain reliability for pharmaceutical manufacturers?
A: By utilizing commercially available starting materials like 1-bromo-2-fluoro-4-(trifluoromethyl)benzene and standard reagents such as n-butyllithium and methyl iodide, the process eliminates dependency on scarce custom building blocks, ensuring consistent raw material availability.
Q: Is this synthetic route suitable for large-scale industrial production?
A: Yes, the process features straightforward workup procedures including extraction, filtration, and crystallization, avoiding complex chromatographic purifications in the final steps, which makes it highly amenable to multi-kilogram and ton-scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Fluoro-3-Methyl-4-(Trifluoromethyl)Benzylamine Hydrochloride Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from patent literature to commercial reality requires more than just chemical knowledge; it demands engineering excellence and rigorous quality assurance. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the promising yields reported in the lab are faithfully reproduced in our manufacturing facilities. We adhere to stringent purity specifications and operate rigorous QC labs equipped with advanced analytical instrumentation to guarantee that every batch of 2-fluoro-3-methyl-4-(trifluoromethyl)benzylamine hydrochloride meets the exacting standards required for clinical and commercial drug substance manufacturing. Our commitment to quality ensures that your downstream processes remain uninterrupted by impurity-related failures.
We invite you to collaborate with us to optimize your supply chain for this critical intermediate. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and timeline. We encourage potential partners to contact us directly to obtain specific COA data from our recent pilot batches and to discuss route feasibility assessments for your unique project needs. Let us leverage our expertise in fluorine chemistry and catalytic processes to become your trusted partner in bringing innovative therapies to market faster and more cost-effectively.