Scalable Synthesis of Benzamide-Substituted Dioxocyclobutene Intermediates for Pharmaceutical Applications
The pharmaceutical industry continuously seeks robust and scalable synthetic routes for complex intermediates, particularly those targeting inflammatory pathways such as CXCR2 antagonists. Patent CN101514168B discloses a highly efficient methodology for the preparation of 2-hydroxy-N,N-dimethyl-3-[[2-[[1(R)-(5-methyl-2-furanyl)propyl]amino]-3,4-dioxo-1-cyclobuten-1-yl]amino]benzamide, referred to herein as Compound of Formula I. This technical disclosure represents a significant advancement in the field of fine chemical synthesis, offering a streamlined approach that addresses common bottlenecks in the production of benzamide-substituted dioxocyclobutene derivatives. The core innovation lies in the strategic convergence of two distinct synthetic fragments: a functionalized salicylamide derivative and a chiral furan-based amine. By leveraging readily available starting materials such as 5-bromo-3-nitrosalicylic acid and 5-methylfurfural, the disclosed process minimizes supply chain risks associated with exotic reagents. Furthermore, the protocol emphasizes operational simplicity, utilizing standard unit operations like hydrogenation, reflux, and crystallization, which are well-understood in industrial settings. For R&D directors and process chemists, this patent provides a blueprint for achieving high purity and yield without relying on labor-intensive purification techniques like column chromatography, thereby setting a new standard for cost reduction in API manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for constructing squaramide-linked pharmacophores often suffer from significant inefficiencies that hinder commercial viability. A primary challenge in conventional methodologies is the handling of unstable intermediates, particularly reduced aniline species derived from nitro-salicylic acid precursors. In many legacy processes, these intermediates must be isolated and purified immediately due to their susceptibility to oxidation or degradation, which introduces additional processing time and increases the risk of yield loss. Moreover, the introduction of chirality into the furan side chain has historically relied on resolution techniques that generate substantial waste or require expensive chiral catalysts that are difficult to recover. Conventional purification strategies frequently depend on silica gel column chromatography to achieve the necessary optical purity for the amine fragment. While effective on a laboratory scale, column chromatography is notoriously difficult to scale up for multi-kilogram or ton-level production due to solvent consumption, throughput limitations, and safety concerns regarding large volumes of organic eluents. These factors collectively contribute to elevated production costs and extended lead times, creating friction for procurement managers seeking reliable pharmaceutical intermediate suppliers who can guarantee consistent quality at competitive prices.
The Novel Approach
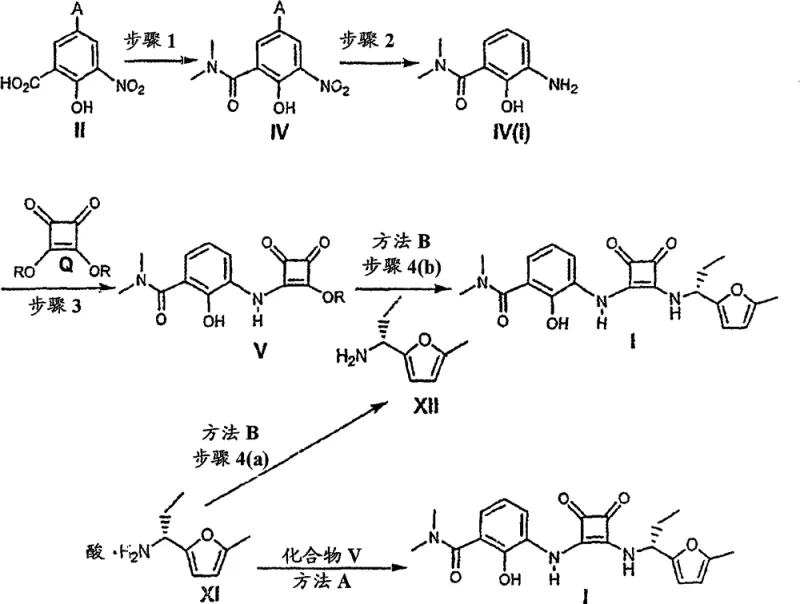
The methodology outlined in CN101514168B overcomes these historical limitations through a series of strategic process optimizations designed for scalability and efficiency. A standout feature of this novel approach is the telescoping of the reduction and coupling steps for the salicylamide fragment. Instead of isolating the unstable aniline intermediate (Formula IV(i)), the process generates it in situ via catalytic hydrogenation and immediately reacts it with diethyl squarate (Formula Q). This "one-pot" strategy not only protects the sensitive amine from degradation but also drastically reduces the number of unit operations, leading to substantial cost savings in terms of labor, energy, and solvent usage. Additionally, the synthesis of the chiral amine fragment employs a chiral pool strategy using R-2-(-)-phenylglycinol, which acts as both a directing group and a resolving agent. Crucially, the final chiral amine is isolated as a stable salt (Formula XI) through crystallization. This shift from chromatographic purification to crystallization is a game-changer for commercial scale-up of complex pharmaceutical intermediates, as it allows for the processing of much larger batches with higher throughput and lower environmental impact. The result is a robust, reproducible process that delivers high-purity intermediates suitable for downstream API synthesis.
Mechanistic Insights into Squaramide Formation and Chiral Induction
The chemical elegance of this synthesis is rooted in the precise control of reactivity and stereochemistry during the formation of the squaramide core and the chiral side chain. The construction of the squaramide linkage involves the nucleophilic attack of the aniline nitrogen (generated from the reduction of the nitro group in Formula IV) onto one of the ester carbonyls of the diethyl squarate (Formula Q). This reaction is typically base-catalyzed, utilizing mild bases such as potassium carbonate or triethylamine to deprotonate the aniline, thereby enhancing its nucleophilicity without promoting hydrolysis of the squarate ester. The reaction conditions are carefully tuned, often maintaining temperatures between 20°C to 50°C, to ensure selective mono-substitution. If the temperature is too high, there is a risk of double substitution or ester hydrolysis, which would compromise the integrity of the squaramide scaffold. The mechanistic pathway ensures that the resulting intermediate (Formula V) retains a reactive ester functionality, poised for the final coupling with the chiral amine. This step-wise assembly allows for rigorous quality control at the intermediate stage, ensuring that any impurities from the initial salicylamide synthesis do not carry over into the final product.
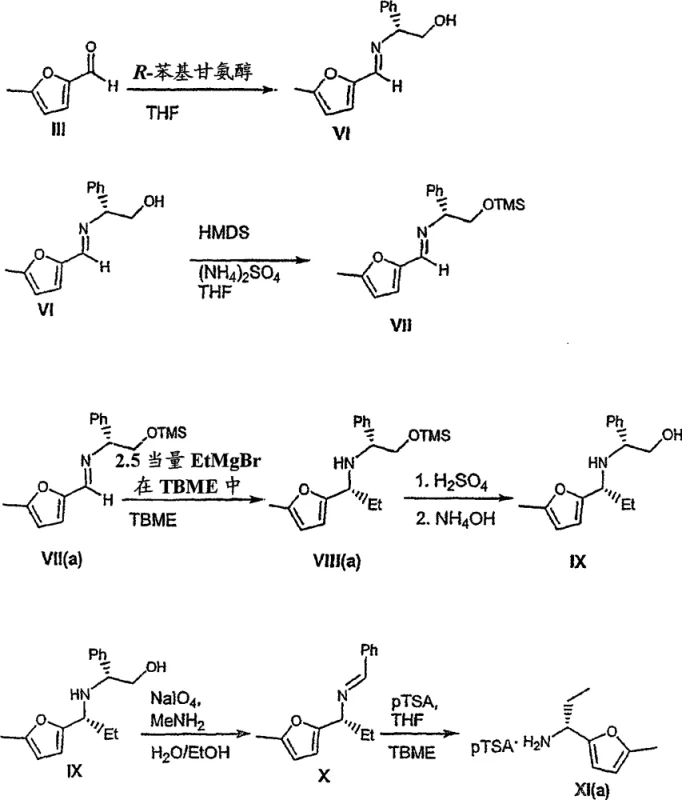
Parallel to the squaramide formation is the sophisticated stereochemical control exercised in the synthesis of the furan amine fragment. The process initiates with the condensation of 5-methylfurfural (Formula III) with R-2-(-)-phenylglycinol to form a chiral imine (Formula VI). This imine serves as a template for the subsequent asymmetric addition of an organometallic reagent, specifically ethylmagnesium bromide. The chiral environment created by the phenylglycinol moiety directs the approach of the Grignard reagent, favoring the formation of one diastereomer over the other. Following the addition, the chiral auxiliary is cleaved under acidic conditions, and the resulting amine is subjected to oxidative cleavage of the amino alcohol side chain using sodium periodate. The final step involves the formation of a salt, typically with p-toluenesulfonic acid, which locks the molecule into a specific crystalline lattice. This crystallization process is thermodynamically driven to exclude the unwanted enantiomer, effectively upgrading the optical purity of the material. This mechanism of chiral induction followed by salt-mediated purification is a powerful tool for ensuring that the final Compound I meets the stringent enantiomeric excess requirements mandated by regulatory bodies for pharmaceutical substances.
How to Synthesize Benzamide-Substituted Dioxocyclobutene Efficiently
The execution of this synthetic route requires careful attention to reaction parameters to maximize yield and purity while maintaining safety standards. The process is divided into the preparation of the squaramide intermediate and the chiral amine, which are then converged in the final step. Operators must ensure that the hydrogenation step is conducted under controlled pressure (typically 100 to 120 psi) to fully reduce the nitro group without over-reducing other sensitive functionalities. Similarly, the Grignard addition must be performed at controlled temperatures (0°C to 35°C) to manage the exotherm and maintain stereoselectivity. The detailed standardized synthetic steps see the guide below.
- Convert 5-bromo-3-nitrosalicylic acid (Formula II) to the corresponding dimethylamide (Formula IV) using thionyl chloride and dimethylamine.
- Hydrogenate Formula IV to the aniline intermediate (Formula IV(i)) and react with diethyl squarate (Formula Q) to form the squaramide intermediate (Formula V).
- Synthesize the chiral furan amine (Formula XI) from 5-methylfurfural using R-phenylglycinol, followed by final coupling with Formula V to yield the target Compound I.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the technical advantages of this patent translate directly into tangible business benefits, primarily centered around cost stability and supply reliability. The reliance on commodity chemicals such as 5-bromo-3-nitrosalicylic acid and 5-methylfurfural means that the raw material supply chain is robust and less susceptible to the volatility often seen with specialized reagents. These starting materials are produced by multiple manufacturers globally, reducing the risk of single-source dependency and ensuring continuous availability even during market fluctuations. Furthermore, the elimination of column chromatography from the purification workflow is a critical factor in driving down the cost of goods sold (COGS). Chromatography is a resource-intensive operation that requires significant amounts of silica gel and solvents, along with specialized equipment and longer cycle times. By replacing this with crystallization, the process becomes inherently more scalable, allowing for larger batch sizes and faster turnaround times. This efficiency gain enables suppliers to offer more competitive pricing structures without compromising on the quality of the high-purity pharmaceutical intermediates delivered to the client.
- Cost Reduction in Manufacturing: The process achieves significant economic optimization by telescoping multiple reaction steps and eliminating the need for chromatographic purification. The in situ generation and reaction of the aniline intermediate remove the costs associated with isolation, drying, and re-dissolution of unstable compounds. Additionally, the use of crystallization for the chiral amine significantly reduces solvent consumption and waste disposal costs compared to traditional column chromatography. These operational efficiencies cumulatively lead to a leaner manufacturing process, allowing for substantial cost savings that can be passed down the supply chain, making the final API more economically viable for the end manufacturer.
- Enhanced Supply Chain Reliability: The synthetic route is designed with supply chain resilience in mind, utilizing starting materials that are commercially available in bulk quantities from established chemical suppliers. The avoidance of exotic or highly specialized catalysts reduces the complexity of the procurement process and mitigates the risk of delays caused by sourcing difficulties. Moreover, the robustness of the crystallization steps ensures that the process can tolerate minor variations in raw material quality without failing, providing a buffer against supply chain disruptions. This reliability is crucial for maintaining consistent production schedules and meeting the demanding delivery timelines required by pharmaceutical clients, ensuring that the reliable pharmaceutical intermediate supplier can fulfill orders without interruption.
- Scalability and Environmental Compliance: From an environmental and regulatory perspective, this process offers a greener alternative to conventional methods. The reduction in solvent usage, particularly the avoidance of large volumes of chromatography eluents, aligns with modern green chemistry principles and helps facilities meet increasingly stringent environmental regulations. The ability to scale the process from kilogram to multi-ton levels using standard reactor equipment (hydrogenators, crystallizers) means that production capacity can be ramped up quickly to meet surges in demand. This scalability ensures that the supply of these critical intermediates can grow in tandem with the clinical and commercial success of the downstream drug, providing a secure long-term supply partner for pharmaceutical development projects.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and supply of these intermediates. The answers are derived from the specific technical disclosures within the patent documentation, ensuring accuracy and relevance for potential partners. Understanding these details is essential for evaluating the feasibility of integrating this technology into existing production pipelines.
Q: What are the key advantages of the synthesis method described in CN101514168B?
A: The method utilizes readily available and inexpensive starting materials like 5-bromo-3-nitrosalicylic acid. It avoids the isolation of unstable reduction intermediates and eliminates the need for column chromatography in the purification of the chiral amine, significantly enhancing scalability and operational safety.
Q: How is chiral purity ensured in the production of the furan amine intermediate?
A: Chiral purity is achieved through the use of R-2-(-)-phenylglycinol as a chiral auxiliary during the imine formation and subsequent Grignard addition. The process further enhances purity by converting the final amine into a stable salt (Formula XI), which can be crystallized to high optical purity.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the process is designed for scalability. Key features such as telescoped reactions (without isolating unstable intermediates), the use of common solvents like ethanol and acetonitrile, and purification via crystallization rather than chromatography make it highly amenable to commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Benzamide-Substituted Dioxocyclobutene Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of having a manufacturing partner who can translate complex patent chemistry into commercial reality. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from pilot plant to full-scale manufacturing. We understand that the synthesis of CXCR2 antagonist intermediates requires precise control over chirality and purity. Therefore, our facilities are equipped with state-of-the-art rigorous QC labs and analytical instruments capable of verifying stringent purity specifications, including chiral HPLC and NMR analysis. We are committed to delivering intermediates that not only meet but exceed the quality standards required for pharmaceutical applications, providing you with the confidence to advance your drug development programs.
We invite you to engage with our technical procurement team to discuss how we can support your specific needs. Whether you require a Customized Cost-Saving Analysis for your current supply chain or need to evaluate the feasibility of this specific synthetic route for your portfolio, we are here to help. Please contact us to request specific COA data and route feasibility assessments tailored to your project milestones. Let us be your partner in bringing innovative therapies to market efficiently and reliably.