Scalable Organocatalytic Synthesis of Chiral Gamma-Amino Acids for Commercial API Production
Introduction to Advanced Chiral Gamma-Amino Acid Synthesis
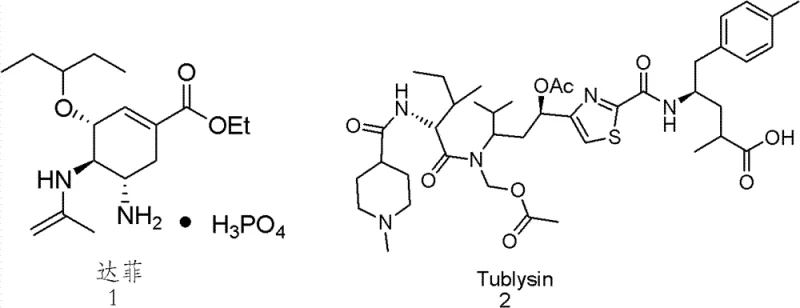
The pharmaceutical industry continuously seeks robust and efficient pathways to access high-value chiral building blocks, particularly for the development of next-generation antiviral and anticancer therapeutics. According to patent CN102249833B, a groundbreaking methodology has been established for the preparation of chiral gamma-amino acids and their derivatives, addressing critical bottlenecks in modern medicinal chemistry. This technology leverages advanced organocatalytic asymmetric cyclization reactions, utilizing chiral carbenes or cinchona alkaloids to induce high levels of stereocontrol during the formation of complex heterocyclic intermediates. The significance of this approach lies in its ability to construct the gamma-amino acid scaffold directly from simple unsaturated acid chlorides and electron-deficient azo or nitroso compounds, bypassing the lengthy and inefficient sequences traditionally associated with amino acid synthesis. By operating under mild conditions, typically around minus twenty degrees Celsius, this process ensures the integrity of sensitive functional groups while delivering products with exceptional enantiomeric purity, often exceeding ninety-nine percent ee. This represents a paradigm shift in how key pharmaceutical intermediates, such as those found in oseltamivir phosphate and tubulysin analogues, are manufactured, offering a streamlined route that aligns perfectly with the rigorous demands of contemporary drug supply chains.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of chiral gamma-amino acids has relied heavily on the homologation of chiral alpha-amino acids, a strategy fraught with significant operational and economic disadvantages for large-scale manufacturing. These conventional routes typically involve multi-step sequences that require the protection and deprotection of various functional groups, leading to substantial material loss and increased waste generation. Furthermore, the reagents employed in these traditional methods are often prohibitively expensive and difficult to source in bulk quantities, creating supply chain vulnerabilities for pharmaceutical producers. The overall yields of these legacy processes are frequently low, necessitating larger reactor volumes and longer production cycles to achieve the same output, which drastically inflates the cost of goods sold. Additionally, many classical approaches rely on stoichiometric amounts of chiral auxiliaries or expensive transition metal complexes, which not only add to the raw material costs but also introduce complex purification challenges to remove trace metal impurities to meet regulatory standards. The cumulative effect of these inefficiencies is a manufacturing process that is rigid, costly, and ill-suited for the rapid scale-up required in the fast-paced environment of generic drug production and active pharmaceutical ingredient supply.
The Novel Approach
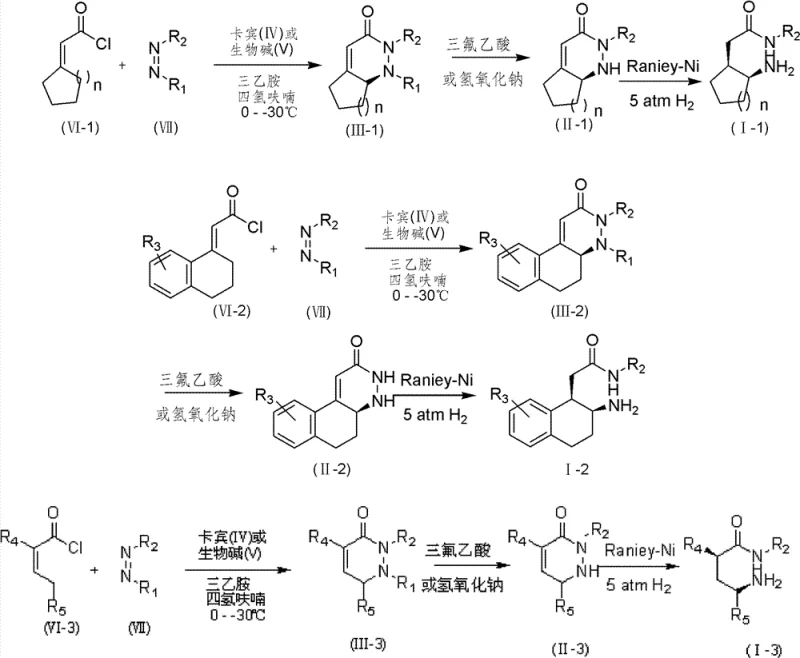
In stark contrast, the novel organocatalytic approach detailed in the patent data offers a direct and highly efficient alternative that fundamentally restructures the synthesis logic. By employing a catalytic amount of a chiral organic small molecule, such as a nitrogen-heterocyclic carbene or a cinchona alkaloid derivative, the reaction achieves asymmetric induction without the need for precious metals in the key bond-forming step. This method facilitates a one-pot cyclization between unsaturated acid chlorides and azo or nitroso compounds to form chiral piperazinones or oxazinones with high diastereoselectivity and enantioselectivity. The subsequent transformation involves a straightforward deprotection followed by a catalytic hydrogenation step using cost-effective catalysts like Raney-Ni or Palladium on Carbon to cleave the nitrogen-nitrogen or nitrogen-oxygen bonds. This streamlined sequence significantly reduces the number of unit operations, minimizes solvent consumption, and simplifies the downstream processing workflow. The ability to generate the chiral center directly during the cyclization event eliminates the need for resolution steps or the use of chiral pool starting materials, thereby unlocking a more flexible and economically viable pathway for producing diverse gamma-amino acid libraries essential for modern drug discovery programs.
Mechanistic Insights into Organocatalytic Asymmetric Cyclization
The core of this technological breakthrough resides in the sophisticated mechanism of the organocatalytic asymmetric cyclization, which dictates the stereochemical outcome of the final product. The reaction initiates with the activation of the electron-deficient azo or nitroso compound by the chiral catalyst, forming a reactive intermediate that is poised for nucleophilic attack. When a chiral carbene catalyst is utilized, it likely forms a transient zwitterionic species with the unsaturated acid chloride, organizing the transition state in a highly ordered manner that favors the formation of one specific enantiomer over the other. Alternatively, when cinchona alkaloids are employed, they act as bifunctional catalysts, simultaneously activating both the electrophile and the nucleophile through hydrogen bonding or ion-pairing interactions. This dual activation lowers the energy barrier for the cyclization while imposing strict steric constraints that ensure the incoming groups approach from the correct face of the molecule. The result is the formation of a chiral piperazinone or oxazinone ring system with precise control over the newly generated stereocenters. This level of control is critical because the biological activity of gamma-amino acid derivatives is often exquisitely sensitive to their three-dimensional configuration, and even minor deviations can lead to inactive or toxic byproducts. The robustness of this catalytic cycle allows it to tolerate a wide range of substituents on the starting materials, providing chemists with the versatility to tune the physicochemical properties of the final amino acid without compromising the stereochemical integrity of the synthesis.
Following the cyclization, the mechanism proceeds through a carefully orchestrated deprotection and reduction sequence that preserves the chirality established in the first step. The removal of protecting groups using trifluoroacetic acid is conducted under mild thermal conditions to prevent racemization, ensuring that the optical purity achieved during catalysis is maintained throughout the process. The subsequent hydrogenation step involves the adsorption of the intermediate onto the surface of the heterogeneous catalyst, where the weak nitrogen-nitrogen or nitrogen-oxygen bonds are selectively cleaved in the presence of hydrogen gas. This step is mechanistically distinct from the initial organocatalytic event but is equally vital for the success of the overall route. The use of Raney-Ni or Pd-C allows for the efficient reduction of the heterocyclic ring to the open-chain gamma-amino acid structure without affecting other sensitive functionalities that might be present on the side chains. The combination of these mechanistic elements creates a cohesive synthetic strategy that is not only theoretically elegant but also practically superior, offering a reliable method for generating high-purity chiral intermediates that meet the stringent quality specifications required for pharmaceutical applications.
How to Synthesize Chiral Gamma-Amino Acid Efficiently
Implementing this synthesis route requires careful attention to reaction parameters to maximize yield and enantiomeric excess, as outlined in the experimental examples of the patent. The process begins with the preparation of the catalyst system, where the chiral carbene precursor or alkaloid is mixed with a base such as cesium carbonate or triethylamine in an appropriate organic solvent like toluene or tetrahydrofuran. Once the catalyst is activated, the electron-deficient azo or nitroso compound is introduced at low temperatures, typically between zero and minus thirty degrees Celsius, to control the exotherm and ensure high selectivity. The unsaturated acid chloride is then added slowly to maintain the reaction concentration within the optimal range, allowing the cyclization to proceed over several hours until the starting materials are fully consumed. After the formation of the chiral heterocyclic intermediate, the reaction mixture is worked up to isolate the protected species, which is then subjected to acidic deprotection. Finally, the deprotected intermediate is dissolved in a solvent compatible with hydrogenation, such as ethanol or ethyl acetate, and treated with the metal catalyst under a hydrogen atmosphere to yield the final gamma-amino acid product. For a detailed breakdown of the specific molar ratios, temperature profiles, and workup procedures tailored to your specific target molecule, please refer to the standardized guide below.
- Perform asymmetric cyclization of unsaturated acid chlorides with electron-deficient azo or nitroso compounds using chiral carbene or alkaloid catalysts at low temperatures.
- Execute deprotection of the resulting chiral piperazinone or oxazinone intermediates using trifluoroacetic acid under controlled conditions.
- Conclude with catalytic hydrogenation using Raney-Ni or Pd-C to cleave nitrogen-nitrogen or nitrogen-oxygen bonds, yielding the final chiral gamma-amino acid.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this organocatalytic technology offers transformative benefits that directly impact the bottom line and operational resilience of pharmaceutical manufacturing. The elimination of expensive transition metal catalysts in the primary stereoselective step represents a major cost reduction opportunity, as it removes the need for sourcing scarce precious metals and the associated analytical costs for residual metal testing. Furthermore, the use of readily available starting materials, such as simple unsaturated acid chlorides and commercially sourced azo compounds, enhances supply chain reliability by reducing dependence on specialized custom synthesis vendors. This accessibility ensures that production schedules are less likely to be disrupted by raw material shortages, providing a more stable foundation for long-term manufacturing planning. The simplified process flow, characterized by fewer reaction steps and easier purification protocols, also translates into significant efficiency gains, allowing facilities to produce larger batches in less time with reduced labor and utility consumption. These factors collectively contribute to a more agile and cost-effective supply chain capable of responding quickly to market demands while maintaining high standards of quality and compliance.
- Cost Reduction in Manufacturing: The substitution of precious metal catalysts with organic small molecules in the key asymmetric step drastically lowers the raw material expenditure and eliminates the complex waste treatment costs associated with heavy metal disposal. Additionally, the high yields and selectivity of the reaction minimize the loss of valuable intermediates, ensuring that a greater proportion of the input materials are converted into saleable product, which substantially improves the overall process economics and reduces the cost per kilogram of the final active pharmaceutical ingredient.
- Enhanced Supply Chain Reliability: By relying on commodity chemicals and robust organocatalysts that are stable and easy to handle, manufacturers can secure a more consistent supply of critical reagents, mitigating the risks associated with geopolitical instability or single-source supplier dependencies. The simplicity of the reaction conditions also means that the process can be easily transferred between different manufacturing sites or contract development and manufacturing organizations without extensive re-optimization, ensuring continuity of supply even in the face of unexpected operational disruptions at a specific facility.
- Scalability and Environmental Compliance: The process is inherently scalable due to its reliance on standard chemical engineering unit operations such as stirred tank reactors and filtration systems, avoiding the need for specialized equipment that might limit batch size. Moreover, the reduction in hazardous waste generation and the avoidance of toxic heavy metals align with increasingly stringent environmental regulations, facilitating smoother regulatory approvals and reducing the environmental footprint of the manufacturing operation, which is a key consideration for sustainable supply chain management in the modern pharmaceutical industry.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this chiral gamma-amino acid synthesis technology, based on the data provided in the patent documentation. These answers are designed to clarify the operational feasibility and strategic value of adopting this method for your specific production needs. Understanding these details is crucial for making informed decisions about process integration and resource allocation. We encourage you to review these insights carefully to assess how this technology can fit into your existing manufacturing portfolio.
Q: What are the primary advantages of this organocatalytic method over traditional homologation?
A: This method eliminates the need for expensive transition metal catalysts in the initial stereoselective step, utilizes readily available starting materials like unsaturated acid chlorides, and achieves significantly higher yields and enantiomeric excess compared to traditional alpha-amino acid homologation routes.
Q: Can this synthesis route be scaled for industrial API manufacturing?
A: Yes, the process is designed for scalability. It employs robust reaction conditions, avoids sensitive reagents in the critical stereochemistry-determining step, and utilizes standard hydrogenation equipment for the final reduction, making it highly suitable for commercial scale-up.
Q: What types of chiral gamma-amino acid derivatives can be produced?
A: The technology supports the synthesis of a diverse range of derivatives, including cyclic and acyclic variants, by varying the unsaturated acid chloride and the azo or nitroso coupling partners, allowing for broad application in drug discovery and development.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral Gamma-Amino Acid Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of accessing high-quality chiral intermediates to drive your drug development programs forward. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial manufacturing is seamless and efficient. We are committed to delivering products that meet stringent purity specifications through our rigorous QC labs, guaranteeing that every batch of chiral gamma-amino acid supplied adheres to the highest international standards. Our expertise in organocatalytic processes allows us to optimize this specific technology for maximum yield and cost-efficiency, providing you with a competitive advantage in the marketplace. Whether you require custom synthesis services or bulk supply of established intermediates, our infrastructure is designed to support your growth and innovation goals with reliability and precision.
We invite you to engage with our technical procurement team to discuss how we can tailor this synthesis route to your specific requirements. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the potential economic benefits of switching to this advanced method. We are ready to provide specific COA data and route feasibility assessments to demonstrate our capability to meet your volume and quality needs. Let us partner with you to optimize your supply chain and accelerate your path to market with confidence and assurance.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →