Advanced Synthetic Route for Sorafenib Intermediates: Enhancing Purity and Scalability for Global Pharma Supply Chains
Advanced Synthetic Route for Sorafenib Intermediates: Enhancing Purity and Scalability for Global Pharma Supply Chains
The global demand for targeted cancer therapies continues to drive innovation in the synthesis of critical kinase inhibitors, with Sorafenib remaining a cornerstone treatment for advanced renal cell carcinoma and hepatocellular carcinoma. As detailed in patent CN108409648B, a novel preparation method for Sorafenib Tosylate related intermediates has been developed that addresses longstanding inefficiencies in traditional manufacturing workflows. This technological breakthrough focuses on the synthesis of 4-(4-aminophenoxy)-N-methyl-2-pyridinecarboxamide, a pivotal building block whose quality directly impacts the safety and efficacy of the final Active Pharmaceutical Ingredient (API). By optimizing reaction sequences and condition parameters, this new approach offers a compelling value proposition for pharmaceutical manufacturers seeking to enhance process reliability while adhering to stringent regulatory standards for impurity profiles.
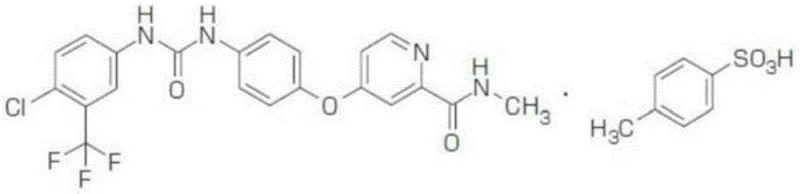
The strategic importance of this intermediate cannot be overstated, as it serves as the nucleophilic partner in the final urea formation step of Sorafenib production. Traditional supply chains have often struggled with batch-to-batch variability and the presence of difficult-to-remove genotoxic impurities. The methodology outlined in the patent data introduces a streamlined pathway that not only improves chemical yield but also fundamentally alters the thermal profile of the synthesis, moving away from energy-intensive heating cycles toward more ambient condition processing. For R&D directors and procurement specialists evaluating potential partners, understanding the mechanistic advantages of this route is essential for securing a stable, cost-effective supply of high-quality oncology intermediates in an increasingly competitive market landscape.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historical manufacturing processes for Sorafenib intermediates have frequently relied on phase transfer catalysis or Williamson ether synthesis protocols that introduce significant operational complexities and environmental burdens. Prior art, such as the methods described in academic literature from Heilongjiang University and various Chinese patents, often necessitates the use of harsh conditions, including prolonged heating at reflux temperatures for up to 12 hours, which drastically increases energy consumption and carbon footprint. Furthermore, these conventional routes are plagued by the formation of tenacious tar-like byproducts during the product isolation phase, which entrap the desired compound and necessitate multiple, yield-depleting recrystallization steps to achieve acceptable purity. The reliance on expensive starting materials like N-methyl-4-chloro-pyridinecarboxamide in some legacy routes further inflates the Cost of Goods Sold (COGS), making the final API less accessible for healthcare systems globally.
The Novel Approach
In stark contrast, the innovative strategy disclosed in patent CN108409648B re-engineers the synthetic sequence to prioritize energy efficiency and operational simplicity. Instead of initiating the synthesis with high-temperature chlorination, this novel approach begins with the formation of the amide bond at the carboxylic acid position, followed by a mild fluorination step conducted at room temperature (20-30°C). This reversal of steps eliminates the need for the 70-80°C heating typically required for pyridine ring functionalization in older methods, resulting in substantial energy savings. The subsequent nucleophilic substitution and catalytic hydrogenation steps are optimized to proceed with high conversion rates, yielding intermediates with purity exceeding 95% directly from the reaction mixture without extensive purification. This streamlined workflow not only accelerates production timelines but also ensures a cleaner impurity profile, which is critical for meeting the rigorous specifications of international regulatory bodies.
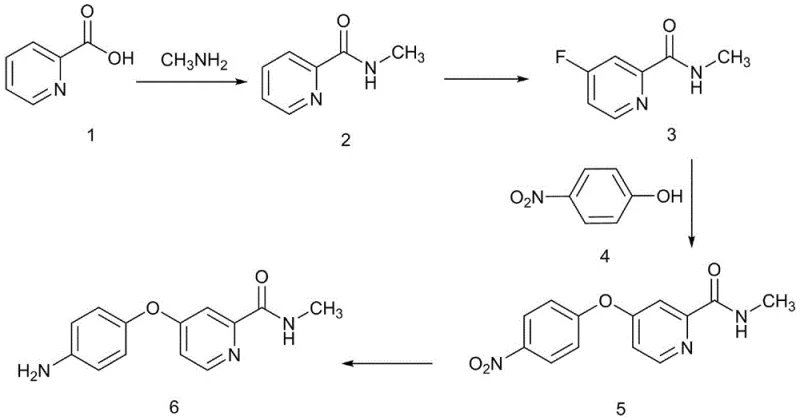
Mechanistic Insights into Fluorination and Nucleophilic Substitution
The core chemical innovation of this process lies in the controlled electrophilic fluorination of the pyridine ring, a transformation that is notoriously difficult to manage due to the high reactivity of fluorine gas. In this patented method, N-methylpicolinamide is subjected to fluorination in the presence of dilute sulfuric acid at a strictly controlled temperature of 25°C. The acidic medium serves to protonate the pyridine nitrogen, effectively deactivating the ring towards unwanted side reactions while directing the electrophilic fluorine species to the desired meta-position relative to the amide group. This precise control prevents over-fluorination and ring degradation, ensuring that the resulting 4-fluoro-N-methylpicolinamide is obtained with a yield of 82-85% and high regioselectivity. The ability to perform this hazardous reaction safely at ambient temperature represents a significant safety engineering achievement, reducing the risk of thermal runaway events that are common in exothermic fluorination processes.
Following fluorination, the synthesis proceeds via a nucleophilic aromatic substitution (SNAr) mechanism where the activated fluoro-pyridine reacts with p-nitrophenol. The presence of the electron-withdrawing amide and the fluorine atom activates the pyridine ring for attack by the phenoxide anion, generated in situ using cesium carbonate as a base. This step is conducted in polar aprotic solvents like DMF at 80°C, facilitating the displacement of the fluoride ion to form the diaryl ether linkage. The choice of cesium carbonate is particularly advantageous as it offers superior solubility and basicity compared to potassium carbonate, driving the reaction to completion within 3 hours with yields reaching 90-92%. The final reduction of the nitro group to the amine is achieved using palladium on carbon (Pd/C) under hydrogen pressure, a robust and scalable catalytic method that avoids the use of stoichiometric metal reductants like iron or tin, thereby simplifying waste treatment and metal removal protocols.
How to Synthesize 4-(4-Aminophenoxy)-N-methyl-2-pyridinecarboxamide Efficiently
The implementation of this synthesis route requires careful attention to reaction parameters, particularly regarding the handling of fluorine gas and the control of hydrogenation pressure. The process is divided into four distinct stages: amidation, fluorination, etherification, and reduction, each designed to maximize throughput while maintaining safety standards. Operators must ensure that the fluorination step is monitored closely via liquid chromatography to prevent over-reaction, stopping the gas flow once the intermediate content exceeds 95%. Similarly, the hydrogenation step utilizes a specific catalyst loading ratio (1:0.0125 mass ratio of substrate to catalyst) to ensure complete conversion without excessive metal contamination. The detailed standardized operating procedures for scaling this chemistry from laboratory benchtop to commercial reactor volumes are critical for maintaining the high purity specifications required for GMP manufacturing.
- Convert 2-picolinic acid to N-methylpicolinamide using thionyl chloride and methylamine in dichloromethane.
- Perform electrophilic fluorination on the pyridine ring at room temperature (20-30°C) using fluorine gas to obtain 4-fluoro-N-methylpicolinamide.
- Execute nucleophilic substitution with p-nitrophenol using cesium carbonate in DMF at 80°C to form the nitro-intermediate.
- Reduce the nitro group to an amino group using Pd/C catalyst and hydrogen pressure to yield the final amine intermediate.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route translates into tangible improvements in supply security and cost structure. By eliminating the high-temperature chlorination step and replacing it with a room-temperature fluorination, the process significantly reduces utility costs associated with heating and cooling large-scale reactors. Furthermore, the avoidance of tar-like byproducts means that filtration and isolation equipment experience less fouling, leading to reduced downtime for cleaning and maintenance between batches. This operational efficiency allows for faster turnaround times and higher asset utilization rates, enabling suppliers to respond more agilely to fluctuations in market demand for Sorafenib APIs without compromising on delivery schedules or product quality.
- Cost Reduction in Manufacturing: The elimination of energy-intensive heating cycles and the use of readily available, cost-effective reagents like fluorine gas and cesium carbonate contribute to a lower overall production cost. Unlike legacy methods that require expensive phase transfer catalysts or specialized chlorinated starting materials, this route utilizes commodity chemicals that are less susceptible to supply chain volatility. The high yield at each step (ranging from 82% to 95%) minimizes raw material waste, ensuring that a greater proportion of input costs are converted into saleable product, thereby improving the gross margin profile for the final API manufacturer.
- Enhanced Supply Chain Reliability: The robustness of this synthetic pathway enhances supply chain resilience by reducing the dependency on complex, multi-step purification processes that are prone to failure. The ability to achieve >95% purity at every intermediate stage means that the risk of batch rejection due to impurity spikes is significantly mitigated. This consistency is vital for maintaining continuous API production lines, as it prevents bottlenecks caused by the need to reprocess off-spec material. Additionally, the use of standard solvents and catalysts ensures that raw material sourcing remains stable, even during periods of global chemical supply constraints.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this method offers distinct advantages by generating less hazardous waste and avoiding the use of heavy metal reductants. The simplified workup procedures reduce the volume of wastewater and organic solvent waste requiring treatment, aligning with increasingly stringent global environmental regulations. The process is inherently scalable, having been designed with industrial production in mind, allowing for seamless transition from pilot plant trials to multi-ton commercial manufacturing without the need for significant process re-engineering or equipment modification.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on how this method outperforms traditional approaches in terms of yield, purity, and operational safety. Understanding these nuances is essential for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: How does this new synthesis route improve energy efficiency compared to conventional methods?
A: Conventional methods often require chlorination at elevated temperatures (70-80°C) prior to amidation. This patented process reverses the order, performing fluorination at room temperature (20-30°C), which significantly reduces thermal energy consumption and operational costs.
Q: What purity levels can be achieved with this manufacturing process?
A: The process is designed for industrial robustness, achieving product purity exceeding 95% at every intermediate step without complex recrystallization. The final intermediate reaches 98.3% purity, minimizing downstream purification burdens for API manufacturers.
Q: Is this method suitable for large-scale commercial production?
A: Yes, the method eliminates tar-like byproducts common in phase transfer catalysis and uses standard solvents like DMF and methanol. The simplified workup and high yields (82-95% per step) make it highly scalable for multi-ton annual production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Sorafenib Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the complexity of modern oncology drug synthesis demands a partner with deep technical expertise and a commitment to quality excellence. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated chemistry required for Sorafenib intermediates is executed with precision and consistency. We operate stringent purity specifications and maintain rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify that every batch meets the exacting standards required for global regulatory submissions. Our capability to implement energy-efficient and environmentally compliant processes like the one described in CN108409648B demonstrates our dedication to sustainable manufacturing practices that benefit both our clients and the broader community.
We invite pharmaceutical companies and contract manufacturing organizations to collaborate with us to leverage these advanced synthetic technologies for their Sorafenib supply chains. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact us today to request specific COA data and route feasibility assessments, allowing us to demonstrate how our optimized manufacturing capabilities can enhance your project's timeline and profitability while ensuring a reliable supply of high-purity pharmaceutical intermediates.