Advanced Synthesis Of Quinazolinone USP7 Inhibitors For Oncology Applications
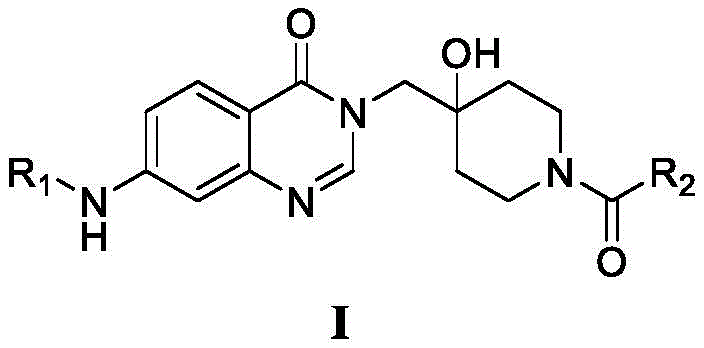
The pharmaceutical industry is continuously seeking robust pathways to develop novel oncology therapeutics, particularly those targeting the ubiquitin-proteasome system. Patent CN112047933B introduces a significant advancement in this field by disclosing a series of quinazolinone-based USP7 inhibitors characterized by General Formula I. These compounds demonstrate remarkable efficacy in inhibiting ubiquitin-specific protease 7, a critical enzyme involved in the regulation of tumor suppressor proteins like p53. The patent details a comprehensive preparation method that is not only chemically elegant but also practically viable for large-scale manufacturing. By leveraging a modular synthetic strategy, the invention allows for the rapid generation of diverse analogues, facilitating extensive structure-activity relationship studies. This technological breakthrough provides a solid foundation for the development of next-generation anticancer medicaments, specifically addressing the urgent need for effective treatments against gastric cancer and other malignancies driven by USP7 dysregulation.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for constructing complex quinazolinone scaffolds often suffer from significant drawbacks that hinder their commercial viability and research utility. Many existing methodologies rely on harsh reaction conditions, such as extremely high temperatures or the use of hazardous reagents, which pose safety risks and increase operational costs. Furthermore, conventional approaches frequently struggle with regioselectivity issues, leading to the formation of unwanted by-products that complicate purification processes and reduce overall yield. The reliance on expensive transition metal catalysts that are difficult to remove to trace levels is another common bottleneck, especially for pharmaceutical intermediates intended for clinical use. These limitations result in prolonged development timelines and inflated production costs, making it challenging for procurement teams to secure reliable supplies of high-purity active pharmaceutical ingredients. Consequently, there is a persistent demand for improved synthetic strategies that can overcome these inefficiencies while maintaining structural diversity.
The Novel Approach
The methodology outlined in patent CN112047933B represents a paradigm shift by offering a streamlined, four-step synthesis that prioritizes efficiency and mild reaction conditions. A key innovation lies in the construction of the spiro-piperidine-quinazolinone core through a sequential cyclization and coupling process that achieves yields as high as 83.7% in critical steps. Unlike traditional methods that might require multiple protection and deprotection cycles, this novel approach utilizes a strategic sequence involving epoxidation, formamide-mediated cyclization, and modern coupling techniques. The use of readily available starting materials, such as substituted benzoic acids and N-Boc-piperidone derivatives, ensures a stable supply chain and reduces raw material costs. Additionally, the final functionalization via Buchwald-Hartwig coupling allows for the precise installation of diverse amine substituents, enabling the fine-tuning of biological activity without compromising the integrity of the core structure. This robust and flexible synthetic platform significantly enhances the feasibility of commercial scale-up.
Mechanistic Insights into The Multi-Step Synthetic Route
The chemical elegance of this synthesis is rooted in its mechanistic precision, particularly during the formation of the quinazolinone ring system. The process initiates with the epoxidation of N-Boc-4-piperidone using trimethylsulfoxonium iodide under basic conditions, generating a reactive spiro-epoxide intermediate. This intermediate then undergoes a nucleophilic attack by the amino group of 2-amino-4-halobenzoic acid in formamide at elevated temperatures, followed by base-mediated ring closure to form the bicyclic quinazolinone structure. This cyclization step is crucial as it establishes the rigid spatial arrangement required for effective binding to the USP7 active site. Subsequent deprotection of the Boc group reveals a secondary amine, which serves as the handle for introducing the R2 side chain via amide bond formation using HATU and triethylamine. This activation strategy ensures high coupling efficiency even with sterically hindered carboxylic acids, minimizing racemization and side reactions. The final step involves a copper-catalyzed C-N cross-coupling reaction, which connects the diverse R1 amine fragments to the chloro-substituted quinazolinone core. This mechanistic pathway ensures that each transformation proceeds with high fidelity, resulting in a final product with an impurity profile that is manageable and consistent with regulatory standards.
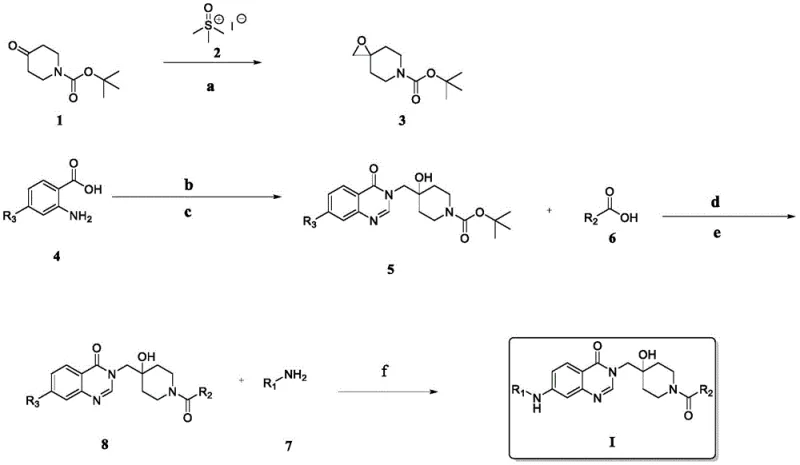
Controlling the impurity profile is paramount in the synthesis of pharmaceutical intermediates, and this patent describes specific measures to achieve high purity. During the amide coupling step, the use of HATU as a coupling reagent is advantageous because it generates soluble urea by-products that are easily removed during aqueous workup, unlike carbodiimide reagents which can form difficult-to-remove urea precipitates. Furthermore, the selection of solvents such as DMF and ethyl acetate allows for effective extraction and crystallization protocols that purge residual starting materials and side products. The patent explicitly mentions purification via column chromatography and recrystallization, indicating a focus on obtaining analytically pure compounds for biological testing. From a quality control perspective, the consistent use of halogenated precursors allows for easy monitoring of reaction progress and product identity using standard spectroscopic techniques. The robustness of the Buchwald-Hartwig coupling condition, utilizing ligands like 2-hydroxyphenylmorpholino ketone, further ensures that the final C-N bond formation does not generate significant amounts of homocoupling by-products. This attention to mechanistic detail translates directly into a cleaner crude product, reducing the burden on downstream purification processes and enhancing the overall economic efficiency of the manufacturing process.
How to Synthesize Quinazolinone USP7 Inhibitors Efficiently
The synthesis of these potent USP7 inhibitors follows a logical progression that balances chemical complexity with operational simplicity. The process begins with the preparation of the spiro-epoxide building block, followed by the assembly of the quinazolinone core, and concludes with the diversification of the side chains. Each step has been optimized to maximize yield and minimize waste, making it an ideal candidate for technology transfer. The detailed standardized synthetic steps见下方的指南 provide a clear roadmap for laboratory execution and subsequent scale-up activities.
- Epoxidation of N-Boc-piperidone using trimethylsulfoxonium iodide and base to form the spiro-epoxide intermediate.
- Cyclization with 2-amino-4-halobenzoic acid in formamide followed by base treatment to construct the quinazolinone core.
- Deprotection and amide coupling with various carboxylic acids using HATU/TEA to introduce the R2 side chain.
- Final Buchwald-Hartwig coupling with amines to install the R1 group, yielding the target USP7 inhibitor.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers tangible benefits that extend beyond mere chemical novelty. The primary advantage lies in the accessibility of raw materials; the key starting materials like 2-amino-4-chlorobenzoic acid and N-Boc-piperidone are commodity chemicals available from multiple global suppliers, mitigating the risk of single-source dependency. This abundance ensures supply continuity even in volatile market conditions. Moreover, the reaction conditions are notably mild, typically operating between room temperature and 150°C, which reduces energy consumption compared to processes requiring cryogenic cooling or extreme heating. The avoidance of exotic or highly toxic reagents simplifies waste management and lowers the environmental compliance burden, aligning with modern green chemistry initiatives. These factors collectively contribute to a more resilient and cost-effective supply chain for producing high-value pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The economic viability of this process is significantly enhanced by the high yields observed in key steps, such as the 83.7% yield in the cyclization reaction. High conversion rates mean less raw material is wasted, directly lowering the cost of goods sold. Furthermore, the use of standard coupling reagents like HATU and common bases like potassium carbonate eliminates the need for proprietary or expensive catalysts that often drive up production costs. The purification methods described, including simple aqueous washes and crystallization, are scalable and do not require specialized equipment, further reducing capital expenditure. By streamlining the synthesis to fewer steps with high efficiency, manufacturers can achieve substantial cost savings in both material and labor, making the final API more competitive in the global market.
- Enhanced Supply Chain Reliability: Supply chain stability is critical for pharmaceutical production, and this synthesis route supports reliability through the use of robust and well-understood chemical transformations. The intermediates generated, such as the Boc-protected quinazolinone, are stable solids that can be stored and transported without degradation, allowing for flexible inventory management. The modularity of the final coupling steps means that different analogues can be produced from a common advanced intermediate, reducing the need to maintain separate stockpiles for every single derivative. This flexibility allows manufacturers to respond quickly to changes in demand or research priorities without disrupting the entire production schedule. Additionally, the tolerance of the reaction conditions to minor variations in parameters ensures consistent batch-to-batch quality, reducing the risk of production failures that could lead to supply shortages.
- Scalability and Environmental Compliance: Scaling chemical processes from the laboratory to commercial production often presents significant challenges, but this route is designed with scalability in mind. The solvents used, such as ethanol, ethyl acetate, and DMF, are widely used in the industry and have established recovery and recycling protocols, minimizing environmental impact. The absence of heavy metal catalysts in the early stages reduces the complexity of metal scavenging steps later in the process, which is a common bottleneck in scale-up. The waste streams generated are primarily organic and aqueous, which can be treated using standard effluent treatment plants. This alignment with environmental regulations facilitates faster regulatory approval and reduces the long-term liability associated with hazardous waste disposal, making it a sustainable choice for long-term commercial manufacturing.
Frequently Asked Questions (FAQ)
Understanding the technical and commercial nuances of this technology is essential for stakeholders evaluating its potential. The following questions address common inquiries regarding the synthesis, activity, and applicability of these quinazolinone derivatives. The answers are derived directly from the experimental data and technical specifications provided in the patent documentation, ensuring accuracy and relevance for decision-makers.
Q: What is the key advantage of this synthesis route?
A: The route utilizes mild reaction conditions and commercially available starting materials, achieving high yields (up to 83.7%) in key cyclization steps without requiring extreme temperatures or pressures.
Q: What is the biological activity of these compounds?
A: The compounds exhibit potent inhibition against USP7 enzyme and show significant anti-proliferative activity against gastric cancer cell lines such as MGC-803 and BGC-823.
Q: Is this process scalable for commercial production?
A: Yes, the synthesis avoids sensitive reagents and uses standard organic solvents like DMF and ethanol, making it highly suitable for scale-up from laboratory to industrial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Quinazolinone USP7 Inhibitor Supplier
The development of USP7 inhibitors represents a frontier in oncology research, and having a partner capable of navigating the complexities of their synthesis is invaluable. NINGBO INNO PHARMCHEM stands ready to support your projects with our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facility is equipped with state-of-the-art rigorous QC labs that ensure every batch meets stringent purity specifications required for preclinical and clinical development. We understand the critical nature of timeline and quality in drug discovery, and our team is dedicated to providing seamless technology transfer and process optimization services. Whether you require custom synthesis of specific analogues or bulk supply of key intermediates, our infrastructure is designed to deliver consistency and reliability.
We invite you to collaborate with us to accelerate your research and development goals. Our technical procurement team is available to discuss your specific requirements and provide a Customized Cost-Saving Analysis tailored to your project volume. We encourage you to reach out to request specific COA data and route feasibility assessments for the quinazolinone USP7 inhibitors described in CN112047933B. By partnering with NINGBO INNO PHARMCHEM, you gain access to a wealth of chemical expertise and manufacturing capacity that can help bring your innovative therapies to patients faster and more efficiently.