Scalable Synthesis of Indole Derivatives for High-Purity Anlotinib Manufacturing
Scalable Synthesis of Indole Derivatives for High-Purity Anlotinib Manufacturing
The pharmaceutical industry continuously seeks robust synthetic pathways for complex kinase inhibitors, particularly for oncology applications where purity standards are stringent. Patent CN115850237A introduces a transformative synthesis method for indole derivatives, specifically targeting the production of Anlotinib intermediates. This technology addresses critical bottlenecks in existing manufacturing processes by eliminating the need for column chromatography purification, a step that often limits throughput and increases operational costs in large-scale API production. By streamlining the conversion of nitro-substituted precursors into the final indole scaffold, this method ensures high yields and superior purity profiles, making it an ideal candidate for reliable pharmaceutical intermediate suppliers aiming to optimize their supply chains. The strategic shift from multi-step purifications to a direct crystallization-based isolation represents a significant leap forward in process chemistry efficiency.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for Anlotinib intermediates, such as those disclosed in CN102159078A, suffer from inherent inefficiencies that hinder industrial scalability. These legacy methods typically involve a four-step sequence starting from 7-benzyloxy-4-chloro-6-methoxyquinoline, which not only results in suboptimal overall yields but also generates significant quantities of stubborn impurities. A major technical hurdle in these conventional pathways is the formation of hydroxylamine byproducts on the nitrogen atom of the indole ring during the penultimate reaction step. These hydroxylamine impurities are chemically similar to the target product, making them exceptionally difficult to remove without resorting to resource-intensive column chromatography. Consequently, manufacturers face increased solvent consumption, longer cycle times, and higher waste generation, all of which negatively impact the cost reduction in API manufacturing and complicate regulatory compliance regarding residual solvents and impurities.
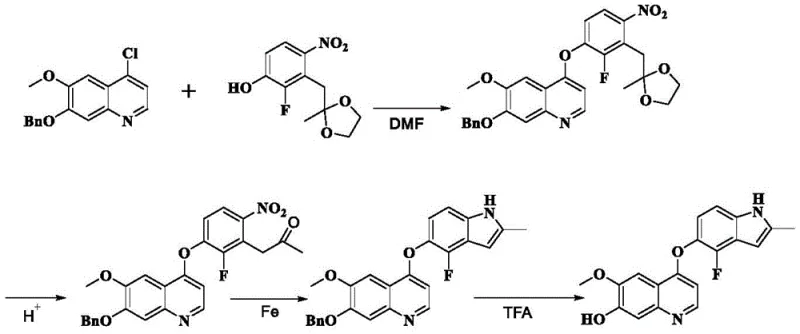
The Novel Approach
The innovative methodology described in the patent fundamentally restructures the synthetic logic by prioritizing the reduction of the nitro group to an amino group prior to the final cyclization event. This strategic reordering of reaction steps effectively bypasses the formation of the problematic N-hydroxy indole species, thereby simplifying the downstream purification landscape. Instead of relying on chromatographic separation, the new process leverages precise control over reaction conditions and solvent systems to achieve high-purity products through simple filtration and crystallization. For instance, the reduction step utilizes transfer hydrogenation with ammonium formate and palladium on carbon, which is both safer and more scalable than high-pressure hydrogenation methods. This approach not only enhances the commercial scale-up of complex pharmaceutical intermediates but also aligns with green chemistry principles by reducing the environmental footprint associated with extensive solvent usage and silica gel waste disposal.
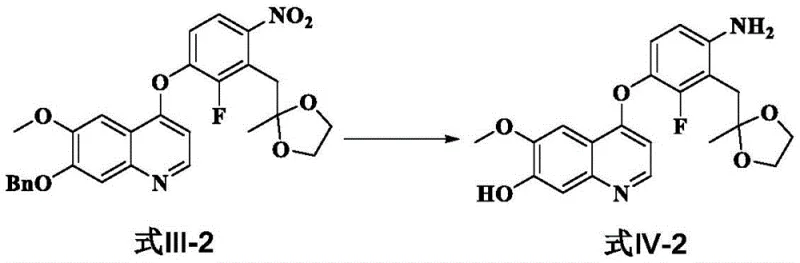
Mechanistic Insights into Nitro Reduction and Cyclization
The core of this technological advancement lies in the selective reduction mechanism employed in the first key transformation. The process utilizes a catalytic transfer hydrogenation system where ammonium formate serves as the hydrogen donor in the presence of a palladium catalyst, typically 10% Pd/C. This reaction proceeds under mild thermal conditions, generally between 30°C and 40°C, which is crucial for preserving the integrity of other sensitive functional groups within the molecule, such as the ether linkages and the quinoline core. The mechanism involves the adsorption of the nitro substrate onto the catalyst surface, followed by the sequential addition of hydrogen atoms derived from the decomposition of ammonium formate. This pathway ensures a clean conversion to the corresponding aniline derivative without over-reduction or side reactions that could compromise the molecular scaffold. Furthermore, the simultaneous removal of benzyl protecting groups during this reduction step adds another layer of efficiency, consolidating two chemical transformations into a single operational unit.
Following the reduction, the subsequent cyclization to form the indole ring is driven by acidic conditions, often utilizing hydrochloric acid in a methanol solvent system. This step facilitates the intramolecular condensation required to close the five-membered heterocyclic ring. The choice of acid and solvent is critical; strong mineral acids like HCl promote the necessary dehydration and aromatization while maintaining the solubility profile needed for effective mixing. From an impurity control perspective, this acidic workup also helps in protonating basic impurities, allowing them to remain in the aqueous phase during the final extraction or filtration steps. The result is a product with a purity exceeding 99%, as demonstrated in the patent examples, which significantly reduces the burden on quality control laboratories and ensures that the material meets the rigorous specifications required for downstream coupling reactions in the final drug substance synthesis.
How to Synthesize Formula V-1 Indole Derivative Efficiently
The practical implementation of this synthesis route is designed for seamless integration into existing cGMP manufacturing facilities. The process begins with the coupling of a chloro-quinoline derivative with a protected nitrophenol, followed by the critical reduction and cyclization steps detailed above. Operators must maintain strict control over temperature gradients and reagent stoichiometry to maximize yield and minimize byproduct formation. The detailed standardized synthesis steps见下方的指南 provide a comprehensive roadmap for executing this chemistry at scale, ensuring reproducibility and consistency across different batch sizes. By adhering to these protocols, production teams can achieve the high purity levels necessary for clinical and commercial supply without the need for specialized chromatographic equipment.
- Couple 7-(benzyloxy)-4-chloro-6-methoxyquinoline with a protected nitrophenol derivative in chlorobenzene using DIPEA at 135°C to form the nitro-intermediate.
- Reduce the nitro group to an amino group using 10% Pd/C and ammonium formate in dichloromethane at 30-40°C, simultaneously removing the benzyl protecting group.
- Cyclize the amino-intermediate into the indole ring by treating with hydrochloric acid in methanol at 60-70°C, followed by neutralization and crystallization.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers tangible benefits that extend beyond mere technical feasibility. The elimination of column chromatography is a game-changer for cost structures, as it removes one of the most expensive and time-consuming unit operations in fine chemical manufacturing. This simplification translates directly into substantial cost savings by reducing the consumption of high-grade solvents and silica gel, while also shortening the overall production lead time. Furthermore, the robustness of the reaction conditions, which utilize common and readily available reagents like ammonium formate and iron powder or palladium catalysts, mitigates the risk of supply disruptions caused by reliance on exotic or scarce materials. This reliability is essential for maintaining continuous production schedules and meeting the demanding delivery timelines of global pharmaceutical clients.
- Cost Reduction in Manufacturing: The primary economic driver of this technology is the complete removal of column chromatography from the purification workflow. In traditional processes, chromatography can account for a significant portion of the total manufacturing cost due to solvent recovery, media replacement, and labor intensity. By switching to a crystallization-based purification strategy, manufacturers can drastically lower their variable costs per kilogram. Additionally, the high yield reported in the patent examples means that less raw material is wasted, further enhancing the overall material efficiency and contributing to a more competitive pricing model for the final intermediate.
- Enhanced Supply Chain Reliability: The use of stable, non-hazardous reagents such as ammonium formate instead of high-pressure hydrogen gas simplifies the safety infrastructure required for production. This reduces the regulatory burden and insurance costs associated with handling hazardous materials. Moreover, the process has been successfully demonstrated in large-scale reactors (200L to 500L), proving its viability for commercial tonnage production. This scalability ensures that suppliers can rapidly ramp up capacity to meet surges in demand without compromising on quality or facing technical bottlenecks that often plague newer, unproven synthetic methods.
- Scalability and Environmental Compliance: From an environmental standpoint, the reduction in solvent usage and the avoidance of silica waste align perfectly with modern sustainability goals. The process generates less hazardous waste, simplifying disposal and lowering the environmental compliance costs for the manufacturing facility. The ability to recycle solvents like dichloromethane and methanol further enhances the green profile of the synthesis. This eco-friendly approach not only appeals to environmentally conscious stakeholders but also future-proofs the supply chain against increasingly stringent environmental regulations in key manufacturing regions.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthesis method. These insights are derived directly from the experimental data and claims presented in the patent documentation, providing a clear understanding of the process capabilities and limitations. Understanding these details is crucial for technical teams evaluating the feasibility of adopting this route for their specific supply chain needs.
Q: How does this method improve impurity profiles compared to prior art?
A: Unlike conventional routes that generate difficult-to-remove hydroxylamine byproducts on the indole nitrogen, this method reduces the nitro group directly to an amine before cyclization, inherently preventing the formation of N-hydroxy impurities and eliminating the need for column chromatography.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the patent explicitly demonstrates scalability in 200L and 500L glass-lined reactors. The avoidance of column chromatography and the use of standard reagents like Pd/C and ammonium formate make it highly adaptable for multi-ton commercial manufacturing.
Q: What are the critical reaction conditions for the reduction step?
A: The reduction of the nitro group is optimally performed using 10% Pd/C catalyst with ammonium formate as the hydrogen source in dichloromethane at a controlled temperature of 30-40°C, ensuring complete conversion while maintaining the integrity of other sensitive functional groups.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Anlotinib Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthetic routes in the development of life-saving oncology therapies. Our team of expert chemists has extensively evaluated the methodology described in CN115850237A and confirmed its potential for delivering high-quality Anlotinib intermediates. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive a consistent and reliable supply of materials. Our state-of-the-art facilities are equipped with the necessary infrastructure to handle the specific reaction conditions required for this synthesis, including large-scale hydrogenation and acidic cyclization capabilities, all while adhering to stringent purity specifications and rigorous QC labs to guarantee product excellence.
We invite potential partners to engage with our technical procurement team to discuss how this advanced synthesis method can be integrated into your supply chain. By leveraging our expertise, you can benefit from a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact us to request specific COA data and route feasibility assessments, allowing you to make informed decisions that drive value and efficiency in your pharmaceutical manufacturing operations.