Advanced Stereoselective Synthesis of Beta-Glycoside Compounds for Commercial API Production
Introduction to Advanced Glycosylation Technology
The global demand for high-purity antiviral agents, particularly nucleoside analogs like Sofosbuvir, has necessitated the development of robust and scalable synthetic routes. Patent CN110981863A introduces a groundbreaking methodology for the preparation of beta-glycoside compounds with exceptional stereoselectivity. This technology addresses one of the most persistent challenges in carbohydrate chemistry: the precise control of anomeric configuration during the glycosylation step. By utilizing a specifically designed imidate donor, referred to as Compound 3, the process achieves a dramatic improvement in beta-selectivity compared to traditional halide or acetate donors. This innovation not only enhances the chemical efficiency of the reaction but also resolves the tedious post-treatment issues often associated with separating alpha and beta isomers. For pharmaceutical manufacturers, this represents a critical advancement in process chemistry, enabling the production of complex nucleoside intermediates with reduced waste and higher overall yield. The strategic implementation of this pathway allows for the reliable supply of key pharmaceutical intermediates required for next-generation Hepatitis C therapies.
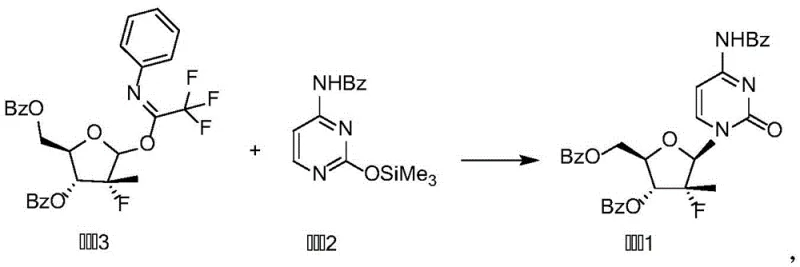
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for constructing the beta-glycosidic linkage in nucleoside analogs have long been plagued by inherent stereochemical ambiguities. Conventional methods often rely on glycosyl halides or simple esters which, under Lewis acid catalysis, frequently yield significant amounts of the thermodynamically stable alpha-anomer alongside the desired beta-product. This lack of selectivity forces manufacturers to employ extensive and costly purification techniques, such as preparative HPLC or multiple recrystallizations, to isolate the active pharmaceutical ingredient. Furthermore, many classical protocols require harsh reaction conditions, including cryogenic temperatures or the use of heavy metal promoters like mercury or tin salts, which pose significant environmental and safety liabilities. The accumulation of these impurities not only drives up the cost of goods sold (COGS) but also complicates regulatory filings due to the stringent requirements for impurity profiling in final drug substances. Consequently, there is an urgent industrial need for a method that intrinsically favors the formation of the beta-configuration without relying on exhaustive downstream processing.
The Novel Approach
The methodology disclosed in the patent circumvents these historical bottlenecks by employing a trifluoroacetimidate derivative (Compound 3) as the glycosyl donor. This novel approach leverages the unique electronic properties of the trifluoromethyl group to activate the anomeric center selectively. As illustrated in the reaction scheme, the coupling of Compound 3 with the silylated base (Compound 2) proceeds smoothly in aromatic solvents like toluene or chlorobenzene. The use of mild organic bases such as triethylamine, DMAP, or DBU facilitates the reaction at moderate temperatures ranging from 60 to 70°C, eliminating the need for cryogenic cooling. This thermal robustness is a significant operational advantage, allowing for easier heat management in large-scale reactors. Most critically, the reaction exhibits a profound preference for the formation of the beta-glycosidic bond, effectively suppressing the generation of alpha-isomers. This intrinsic selectivity transforms the purification workflow from a complex separation challenge into a straightforward filtration and washing procedure, thereby streamlining the entire manufacturing campaign for high-purity pharmaceutical intermediates.
Mechanistic Insights into Trifluoroacetimidate-Mediated Glycosylation
The superior performance of this synthetic route can be attributed to the specific mechanistic pathway enabled by the trifluoroacetimidate functionality. In the presence of a base or mild Lewis acid, the nitrogen atom of the imidate group coordinates with the catalyst, facilitating the departure of the leaving group and generating a reactive oxocarbenium ion intermediate. The electron-withdrawing nature of the trifluoromethyl group stabilizes the transition state and influences the conformational equilibrium of the sugar ring. Unlike bulky trichloroacetimidates, the trifluoro variant offers a distinct balance of reactivity and stability that is particularly well-suited for sensitive nucleoside bases. The neighboring group participation from the C-2 benzoyl protecting group further directs the incoming nucleophile (the silylated base) to attack from the beta-face, ensuring high stereocontrol. This concerted mechanism minimizes the lifetime of the free oxocarbenium ion, reducing the opportunity for anomerization and side reactions. Understanding this mechanistic nuance is vital for process chemists aiming to replicate these results on a commercial scale, as it highlights the importance of maintaining strict stoichiometric control and solvent dryness to preserve the integrity of the reactive donor.
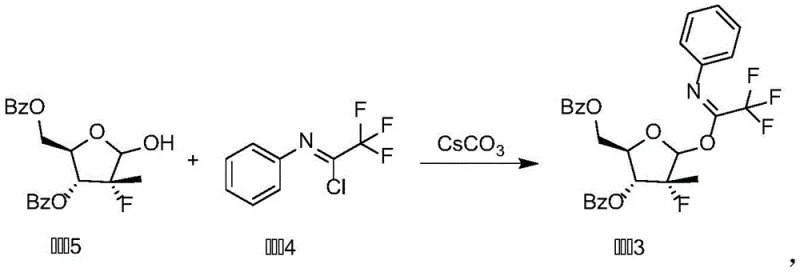
In addition to the coupling step, the preparation of the donor itself (Compound 3) is engineered for high fidelity. The synthesis involves the reaction of a protected sugar alcohol with N-phenyl-trifluoroacetimidoyl chloride in the presence of cesium carbonate. The use of cesium carbonate as a base is particularly advantageous due to its mild basicity and high solubility in organic media, which prevents the degradation of acid-sensitive protecting groups on the sugar scaffold. Conducting this reaction at low temperatures (0-10°C) ensures that the formation of the imidate linkage is kinetically controlled, preventing elimination side reactions. The resulting Compound 3 is a stable, isolable solid that can be purified to homogeneity before being used in the subsequent glycosylation step. This modular approach, where the donor is pre-formed and characterized, adds a layer of quality control to the process, ensuring that every batch of the final nucleoside starts with a verified high-quality building block. Such rigorous control over the starting materials is essential for maintaining consistent impurity profiles in the final API.
How to Synthesize Sofosbuvir Intermediate Efficiently
Implementing this patented technology requires adherence to specific operational parameters to maximize yield and stereoselectivity. The process is divided into two distinct stages: the activation of the sugar moiety and the subsequent coupling with the heterocyclic base. Operators must ensure that all solvents are anhydrous and that the reaction vessels are maintained under an inert atmosphere to prevent hydrolysis of the sensitive imidate donor. The temperature profiles specified in the patent, particularly the 60-70°C range for the coupling reaction, should be strictly monitored to avoid thermal decomposition while ensuring complete conversion. Detailed standardized operating procedures for each unit operation, from the initial mixing of reagents to the final crystallization, are critical for technology transfer. For a comprehensive breakdown of the exact reagent quantities, addition rates, and workup protocols, please refer to the step-by-step synthesis guide below.
- Prepare the glycosyl donor (Compound 3) by reacting the protected sugar alcohol with N-phenyl-trifluoroacetimidoyl chloride using cesium carbonate in dichloromethane at 0-10°C.
- Couple Compound 3 with the silylated pyrimidine base (Compound 2) in toluene or chlorobenzene using an organic base like triethylamine or DBU at 60-70°C.
- Isolate the final beta-glycoside product (Compound 1) via filtration, aqueous washing, and recrystallization from methanol/dichloromethane to achieve >99% purity.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this stereoselective glycosylation method offers substantial benefits for procurement managers and supply chain directors focused on cost optimization and reliability. The primary value driver is the drastic simplification of the purification process. By achieving high beta-selectivity directly in the reactor, the need for expensive chromatographic separations is largely eliminated. This reduction in downstream processing translates directly into lower solvent consumption, reduced waste disposal costs, and shorter cycle times per batch. Furthermore, the use of commodity chemicals such as toluene, chlorobenzene, and triethylamine ensures that raw material sourcing remains stable and cost-effective, insulating the supply chain from volatility associated with exotic reagents. The robustness of the reaction conditions also implies a lower risk of batch failure, enhancing the overall reliability of supply for critical antiviral intermediates.
- Cost Reduction in Manufacturing: The elimination of complex purification steps significantly lowers the operational expenditure associated with producing nucleoside analogs. Traditional methods often require multiple passes through silica columns or extensive recrystallization sequences to remove alpha-isomers, which consumes vast quantities of solvents and labor. By contrast, this novel route allows for product isolation via simple filtration and washing, drastically cutting down on utility usage and waste treatment fees. Additionally, the high yield and purity reduce the amount of starting material required per kilogram of final product, optimizing the overall material balance and driving down the cost of goods sold.
- Enhanced Supply Chain Reliability: The reliance on stable, shelf-solid intermediates like Compound 3 mitigates the risks associated with unstable reagents that require cold chain logistics or immediate use. The ability to synthesize and store the glycosyl donor separately allows for better inventory management and production scheduling flexibility. Moreover, the use of common organic solvents and bases means that procurement teams are not dependent on single-source suppliers for specialized chemicals. This diversification of the supply base ensures continuity of operations even in the face of market disruptions, making the manufacturing process more resilient and predictable for long-term contracts.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, utilizing reaction conditions that are easily manageable in large stainless steel reactors. The absence of heavy metal catalysts removes the need for costly and environmentally burdensome metal scavenging steps, simplifying regulatory compliance and reducing the environmental footprint of the manufacturing site. The moderate temperatures and atmospheric pressure operations further enhance safety profiles, lowering insurance premiums and facility maintenance costs. This alignment with green chemistry principles not only satisfies corporate sustainability goals but also future-proofs the manufacturing asset against tightening environmental regulations.
Frequently Asked Questions (FAQ)
To assist technical decision-makers in evaluating this technology, we have compiled answers to common inquiries regarding the practical application of this patent. These insights are derived directly from the experimental data and technical specifications provided in the intellectual property documentation. Understanding these nuances is crucial for assessing the feasibility of integrating this route into existing production lines. The following section addresses key concerns regarding selectivity, solvent compatibility, and quality metrics.
Q: What is the primary advantage of using Compound 3 in this synthesis?
A: Compound 3 acts as a highly reactive glycosyl donor that significantly improves the stereoselectivity of the beta-glycosidic bond formation, minimizing the formation of unwanted alpha-anomers and simplifying downstream purification.
Q: Which solvents are recommended for the coupling reaction?
A: The patent specifies the use of aromatic organic solvents such as toluene or chlorobenzene, which provide optimal solubility and reaction kinetics for the coupling of the imidate donor with the silylated base.
Q: What purity levels can be achieved with this method?
A: Experimental examples in the patent demonstrate that the final Compound 1 can be isolated with HPLC purity exceeding 99.5%, reaching up to 99.88% when optimized bases like DBU are utilized.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Sofosbuvir Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient synthetic routes in the competitive landscape of antiviral drug manufacturing. Our team of expert process chemists has thoroughly analyzed the technology disclosed in CN110981863A and is fully equipped to implement this high-stereoselective glycosylation method on a commercial scale. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to pilot plant is seamless and risk-mitigated. Our state-of-the-art facilities are designed to handle the specific solvent systems and temperature controls required for this chemistry, guaranteeing consistent output that meets stringent purity specifications. With our rigorous QC labs and commitment to quality assurance, we deliver Sofosbuvir intermediates that exceed industry standards for impurity profiles and stereochemical purity.
We invite potential partners to engage with our technical procurement team to discuss how this advanced synthesis route can optimize your supply chain. By leveraging our manufacturing capabilities, you can secure a stable source of high-quality intermediates while achieving significant operational efficiencies. We encourage you to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. Contact us today to obtain specific COA data and route feasibility assessments, and let us demonstrate how our expertise can drive value for your organization.