Scalable Synthesis of 4'-Norepipodophyllotoxin Derivatives for Next-Generation Antitumor Therapeutics
Scalable Synthesis of 4'-Norepipodophyllotoxin Derivatives for Next-Generation Antitumor Therapeutics
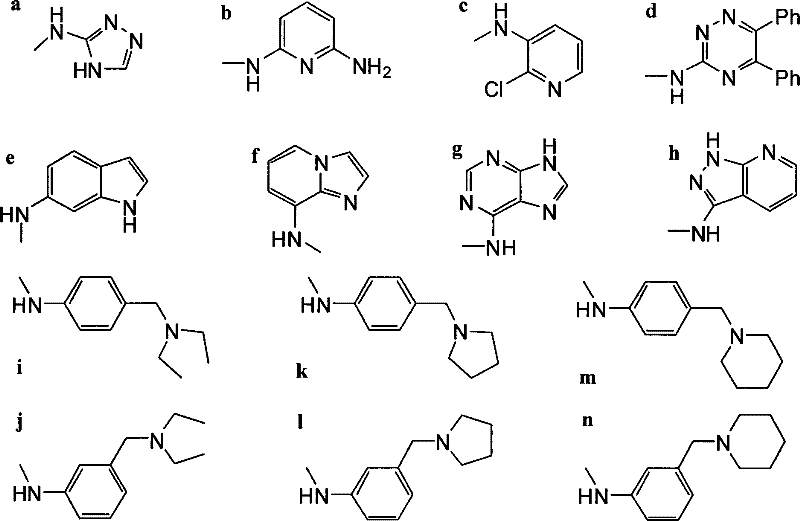
The pharmaceutical industry continuously seeks robust synthetic routes for high-value antitumor intermediates that can overcome the limitations of existing therapies. A pivotal advancement in this domain is detailed in patent CN101074233B, which outlines a novel method for preparing 4'-norepipodophyllotoxin derivatives (compounds IIIa-n). This technology represents a significant leap forward in medicinal chemistry, specifically addressing the critical challenge of drug resistance in cancer treatment. By modifying the C-4 position of the etoposide scaffold through a highly controlled nucleophilic substitution, this process yields a library of compounds with enhanced biological activity. For R&D directors and procurement specialists alike, understanding the mechanistic elegance and commercial viability of this pathway is essential for securing a reliable pharmaceutical intermediates supplier capable of delivering next-generation oncology building blocks.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional methods for modifying podophyllotoxin and its derivatives often suffer from severe stereochemical instability, particularly at the C-4 position. When attempting to introduce amino groups using classical activation strategies, chemists frequently encounter unwanted epimerization, leading to the formation of inactive or toxic isomers that complicate purification and reduce overall yield. Furthermore, conventional routes often require harsh reaction conditions, such as strong acids or elevated temperatures, which can trigger demethylation at the 4'-position, thereby destroying the pharmacophore essential for Topo II inhibition. These side reactions not only diminish the therapeutic potential of the final product but also create complex impurity profiles that are difficult and costly to remove during downstream processing. Consequently, the scalability of these older methods is severely restricted, making them unsuitable for the rigorous demands of modern commercial scale-up of complex pharmaceutical intermediates.
The Novel Approach
In stark contrast, the methodology described in patent CN101074233B offers a streamlined and highly selective alternative that circumvents these historical pitfalls. The process initiates with the conversion of etoposide into a 4-iodo intermediate using sodium iodide and trimethylchlorosilane in dichloromethane at ambient temperature, a mild activation step that preserves the integrity of the sensitive glycosidic bond. Crucially, the subsequent substitution reaction employs barium carbonate to buffer the system at a neutral pH of 7-8, effectively suppressing the acid-catalyzed epimerization that plagues other methods. This strategic use of mild bases allows for the direct introduction of diverse amino compounds, ranging from heterocyclic amines to chain-substituted anilines, with exceptional stereocontrol. The result is a versatile platform for generating high-purity 4'-norepipodophyllotoxin derivatives with yields reaching up to 85% in specific examples, demonstrating clear superiority over legacy synthetic routes.
Mechanistic Insights into TMSCl-Mediated Iodination and Substitution
The core of this synthetic innovation lies in the precise activation of the C-4 hydroxyl group without compromising the stereochemical configuration of the lignan skeleton. The reaction begins with the in situ generation of a reactive silyl species via the interaction of trimethylchlorosilane (TMSCl) with the substrate, which facilitates the displacement of the hydroxyl group by iodide ions from sodium iodide. This transformation proceeds through an SN2-like mechanism that inherently favors inversion or retention depending on the specific conditions, but in this optimized protocol, the conditions are tuned to maintain the desired 4β-configuration. The use of dichloromethane as the solvent ensures excellent solubility of the lipophilic etoposide starting material while providing a non-protic environment that minimizes side reactions. This initial iodination step is critical because the iodine atom serves as an excellent leaving group for the subsequent nucleophilic attack, far superior to the original hydroxyl group or typical sulfonate esters which might require more forcing conditions to displace.
Following the formation of the 4-iodo intermediate, the addition of barium carbonate serves a dual purpose as both a base and a scavenger for the hydrogen iodide generated during the substitution. By maintaining the reaction mixture at a strictly controlled pH of 7-8, the protocol prevents the formation of oxocarbenium ions that would otherwise lead to racemization at the C-4 center. When the amine nucleophile attacks the C-4 position, the presence of the bulky 4'-demethylated scaffold directs the approach of the incoming amine, ensuring high diastereoselectivity. This mechanistic finesse is what allows for the successful incorporation of sterically demanding groups, such as the piperidinylmethyl-anilino moieties found in compounds IIIm and IIIn, without degrading the core structure. For process chemists, this level of control translates directly into reduced purification burdens and higher throughput in the manufacturing of these potent antitumor agents.
How to Synthesize 4'-Norepipodophyllotoxin Derivatives Efficiently
Implementing this synthesis requires careful attention to the stoichiometry of the iodination reagents and the precise timing of the base addition to ensure optimal conversion. The process is designed to be operationally simple, avoiding the need for cryogenic temperatures or exotic catalysts, which makes it highly attractive for transfer from the laboratory to pilot plant scales. Operators should monitor the reaction progress closely, particularly during the substitution phase, to ensure complete consumption of the iodo-intermediate before workup. The detailed standardized synthesis steps, including specific molar ratios and workup procedures for various R-group substitutions, are outlined in the guide below to facilitate immediate adoption by your technical team.
- React etoposide with sodium iodide and trimethylchlorosilane in dichloromethane at room temperature to form the 4-iodo intermediate.
- Add barium carbonate and triethylamine to adjust pH to 7-8, preventing epimerization.
- Introduce the specific amino compound and stir at room temperature for 8-10 hours to yield the target derivative.
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain perspective, this synthetic route offers substantial benefits by leveraging widely available starting materials and minimizing the need for specialized reagents. The reliance on etoposide as the primary feedstock ensures a stable supply chain, as it is a mature commodity in the pharmaceutical market, reducing the risk of raw material shortages. Furthermore, the elimination of harsh acidic conditions and high-temperature steps significantly lowers the energy consumption and safety risks associated with the manufacturing process. This operational simplicity allows for faster batch turnover times and reduces the burden on waste treatment facilities, aligning with modern green chemistry principles and environmental compliance standards.
- Cost Reduction in Manufacturing: The avoidance of expensive transition metal catalysts and the use of inexpensive reagents like sodium iodide and barium carbonate drastically reduces the bill of materials for each batch. By preventing the formation of difficult-to-separate epimers, the process minimizes the loss of valuable material during purification, leading to significant overall cost savings per kilogram of active intermediate produced. Additionally, the ability to run reactions at room temperature eliminates the capital and operational expenditures associated with heating or cooling large-scale reactors, further enhancing the economic viability of the process.
- Enhanced Supply Chain Reliability: Since the synthesis does not depend on proprietary or hard-to-source catalysts, procurement teams can secure multiple vendors for the necessary reagents, mitigating single-source risks. The robustness of the reaction conditions means that the process is less susceptible to minor fluctuations in utility supplies or environmental conditions, ensuring consistent delivery schedules for downstream API manufacturers. This reliability is critical for maintaining continuous production lines for life-saving oncology medications where interruptions can have severe consequences.
- Scalability and Environmental Compliance: The simplified workup procedure, which primarily involves filtration and chromatography, generates less hazardous waste compared to traditional methods that often require extensive aqueous washes with strong acids or bases. This reduction in effluent complexity simplifies wastewater treatment and lowers disposal costs, making the process more sustainable and compliant with increasingly stringent environmental regulations. The high yields reported in the patent examples suggest that the process can be scaled from gram to multi-ton quantities without significant loss of efficiency, supporting long-term commercial growth.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these 4'-norepipodophyllotoxin derivatives. These insights are derived directly from the experimental data and structural analysis provided in the patent literature to assist decision-makers in evaluating the technology. Understanding these nuances is vital for assessing the feasibility of integrating these intermediates into your current drug development pipeline.
Q: How does this synthesis method prevent stereochemical degradation?
A: The process utilizes barium carbonate as a mild base scavenger during the substitution step, which effectively maintains the reaction system at pH 7-8. This specific pH control prevents the acid-catalyzed epimerization at the C-4 position that typically plagues traditional podophyllotoxin modifications.
Q: What is the cytotoxic advantage of these derivatives over etoposide?
A: Preliminary screening indicates that most derivatives, particularly those with chain substituents like IIIk-n, exhibit significantly higher cytotoxic activity against etoposide-resistant cell lines (KB-R), with potency improvements ranging from 10 to 1000 times compared to the parent compound.
Q: Are the starting materials commercially viable for large-scale production?
A: Yes, the synthesis starts directly from etoposide, a widely available commercial API, and utilizes common reagents such as sodium iodide and trimethylchlorosilane, ensuring a robust and cost-effective supply chain for industrial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 4'-Norepipodophyllotoxin Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the synthetic route described in CN101074233B for developing superior antitumor therapies. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition smoothly from benchtop discovery to full-scale manufacturing. Our facility is equipped with rigorous QC labs and adheres to stringent purity specifications, guaranteeing that every batch of 4'-norepipodophyllotoxin derivatives meets the highest international standards for pharmaceutical intermediates.
We invite you to collaborate with us to optimize your supply chain and accelerate your time to market. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our expertise can enhance your production efficiency and product quality.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →