Scalable Synthesis of High-B-Ring Berberine Derivatives for Oncology Applications
Introduction to Novel IDO1/TDO Inhibitory Scaffolds
The landscape of oncology drug discovery is rapidly evolving, with a significant focus shifting towards immunomodulatory agents that can reverse tumor-induced immune suppression. A pivotal development in this sector is detailed in patent CN114736202A, which discloses a series of novel berberine derivatives exhibiting potent inhibitory activity against Indoleamine 2,3-dioxygenase 1 (IDO1) and Tryptophan 2,3-dioxygenase (TDO). These enzymes are critical checkpoints in the kynurenine pathway, and their overexpression in malignant tumors facilitates immune escape by depleting tryptophan levels in the microenvironment. The innovation lies not merely in minor structural tweaks but in the fundamental reconstruction of the berberine core, specifically expanding the B-ring into a seven-membered heterocyclic system containing oxygen, nitrogen, or sulfur atoms. This structural modification addresses the historical limitations of natural berberine, such as poor bioavailability and limited target specificity, offering a new avenue for developing high-purity pharmaceutical intermediates.
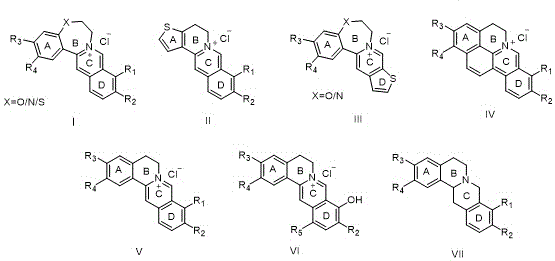
The patent outlines a comprehensive library of compounds, designated as structures I through VII, where the variability in the A and D rings allows for fine-tuning of electronic and steric properties. For R&D directors evaluating new leads, the inclusion of thiophene rings in the A or D positions represents a strategic move to enhance metabolic stability and lipophilicity. Furthermore, the introduction of hydroxyethyl groups via propylene oxide adds a handle for further conjugation or solubility enhancement. As a reliable pharmaceutical intermediate supplier, understanding these structural nuances is essential for anticipating the purification challenges and stability profiles of these complex alkaloids during scale-up.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the modification of berberine has been constrained by the rigidity of its tetracyclic isoquinoline skeleton. Conventional semi-synthetic approaches often rely on harsh demethylation conditions or non-selective alkylation of the quaternary nitrogen, which frequently result in complex mixtures of regioisomers that are difficult to separate. Moreover, extracting sufficient quantities of natural berberine from plant sources like Coptis chinensis to serve as a starting material for extensive medicinal chemistry campaigns introduces supply chain volatility and batch-to-batch variability in impurity profiles. The planar nature of the native molecule also limits its ability to fit into the deeper, more specific pockets of the IDO1 heme domain, leading to moderate potency that often requires high dosing in vivo. These factors collectively increase the cost of goods sold (COGS) and extend the timeline for lead optimization, posing significant risks for procurement managers aiming to secure cost reduction in API manufacturing.
The Novel Approach
The methodology presented in the patent circumvents these bottlenecks by employing a bottom-up synthetic strategy centered on C-H activation and oxidative cyclization. Instead of modifying the natural product, the core isoquinoline scaffold is constructed de novo from simpler, commercially available precursors. This approach allows for the precise installation of the expanded seven-membered B-ring prior to the final cyclization step, ensuring that the heteroatom (O, N, or S) is incorporated with high regioselectivity. For instance, the synthesis of the 5-oxahomoB-ring derivative (Ber-2) utilizes a cobalt-catalyzed coupling of an amide intermediate with a terminal alkyne, followed by a nucleophilic displacement with ethylene glycol. This route avoids the use of expensive chiral catalysts or cryogenic conditions typically associated with complex alkaloid synthesis, thereby streamlining the process for commercial scale-up of complex pharmaceutical intermediates.
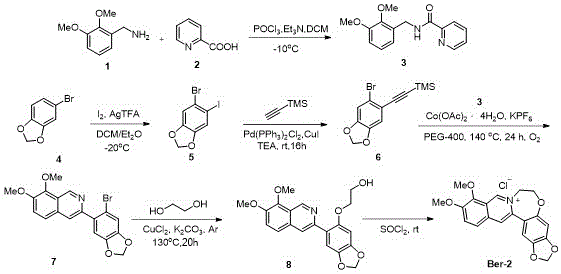
Furthermore, the versatility of this synthetic platform is demonstrated by its ability to accommodate diverse substituents without requiring a complete redesign of the reaction sequence. Whether introducing a methylenedioxy group on the D-ring or a thiophene moiety on the A-ring, the core steps of acylation, halogenation, and metal-catalyzed coupling remain consistent. This modularity is a critical advantage for supply chain heads concerned with reducing lead time for high-purity pharmaceutical intermediates, as it allows for the rapid generation of analog libraries using a standardized set of unit operations. The final ring-closing step using thionyl chloride is a robust, high-yielding transformation that ensures the formation of the quaternary ammonium salt, which is crucial for the biological activity of the final inhibitor.
Mechanistic Insights into Co-Catalyzed Oxidative Cyclization
The heart of this synthetic innovation is the cobalt-catalyzed C-H activation and cyclization step, which constructs the central isoquinoline core while simultaneously establishing the connectivity for the expanded B-ring. In the synthesis of compound 7, a key precursor, the reaction proceeds between an N-(2,3-dimethoxybenzyl)picolinamide and a substituted phenylacetylene in the presence of Co(OAc)2·4H2O and KPF6 within a PEG-400 solvent system at 140 °C. The mechanism likely involves the coordination of the cobalt center to the pyridine nitrogen of the picolinamide directing group, which facilitates the activation of the ortho-C-H bond on the benzyl ring. This metallacycle intermediate then undergoes migratory insertion of the alkyne, followed by reductive elimination to form the new C-C bond. The use of PEG-400 as a solvent is particularly noteworthy; it acts not only as a reaction medium but potentially stabilizes the cationic cobalt species, enhancing the turnover number and allowing the reaction to proceed under aerobic conditions without the need for inert gas protection beyond the initial purge.
Impurity control in this step is managed through careful optimization of the oxidant and temperature. The patent specifies heating the mixture in an oil bath at 140 °C for 24 hours, a condition that ensures complete consumption of the starting alkyne while minimizing the formation of homocoupling byproducts (Glaser coupling). Following the reaction, the workup involves extraction with diethyl ether and purification via silica gel column chromatography using a petroleum ether/ethyl acetate gradient. This rigorous purification protocol is essential for removing trace cobalt residues, which could otherwise catalyze degradation pathways in the final drug substance. For R&D teams, understanding that the KPF6 serves to precipitate the cationic intermediate as a stable hexafluorophosphate salt provides a valuable handle for isolation, significantly improving the overall yield and purity of the intermediate before it proceeds to the final cyclization steps.
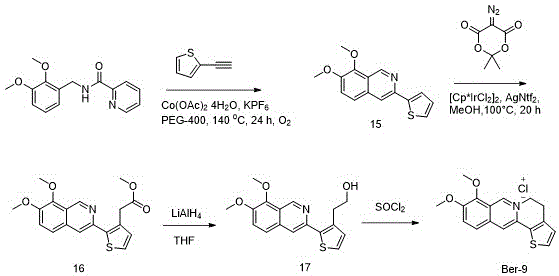
In addition to the cobalt pathway, the patent also explores iridium-catalyzed C-H functionalization for the synthesis of thiophene-containing derivatives like Ber-9. Here, a [Cp*IrCl2]2 catalyst is employed in conjunction with a diazo compound to functionalize the isoquinoline ring. This demonstrates the breadth of the technology platform, offering alternative routes for substrates that may be sensitive to cobalt conditions. The ability to switch between first-row (Co) and second-row (Ir) transition metals provides a strategic fallback for process chemists aiming to optimize cost versus performance. The subsequent reduction of the ester moiety using LiAlH4 and cyclization with SOCl2 showcases a classic yet effective sequence for generating the final quaternary ammonium chloride salt, ensuring that the final product meets the stringent purity specifications required for preclinical toxicology studies.
How to Synthesize High-B-Ring Berberine Derivatives Efficiently
The synthesis of these potent IDO1/TDO inhibitors follows a logical progression of bond-forming reactions that prioritize atom economy and operational simplicity. The process begins with the activation of simple benzylamines and proceeds through well-defined coupling and cyclization events. While the specific stoichiometry and conditions vary slightly depending on the target heteroatom (O, N, or S), the overarching workflow remains consistent, making it highly amenable to technology transfer. For laboratory personnel tasked with replicating these results, attention to detail in the purification of the alkyne intermediates is paramount, as residual halides can poison the cobalt catalyst in the subsequent step. The detailed standardized synthesis steps see the guide below for a breakdown of the critical parameters.
- Preparation of amide intermediates via acylation of benzylamines with picolinic acid derivatives using POCl3.
- Halogenation of bromobenzene derivatives followed by Sonogashira coupling to introduce terminal alkynes.
- Cobalt-catalyzed oxidative C-H activation and cyclization in PEG-400 at elevated temperatures to form the isoquinoline core.
- Final ring closure via nucleophilic substitution with diols or amino-alcohols followed by treatment with thionyl chloride.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the synthetic route described in patent CN114736202A offers distinct advantages over traditional extraction-based methods or complex total syntheses of related alkaloids. The reliance on commodity chemicals such as bromobenzenes, benzylamines, and acetylenes means that the raw material supply chain is robust and less susceptible to the geopolitical or agricultural fluctuations that affect natural product sourcing. This stability is a key factor for procurement managers looking to secure long-term contracts for critical oncology intermediates. Furthermore, the elimination of expensive noble metal catalysts in the primary cyclization step (using Cobalt instead of Palladium or Rhodium where possible) contributes to a significant reduction in the overall cost of goods, aligning with the industry-wide drive for cost reduction in API manufacturing.
- Cost Reduction in Manufacturing: The process utilizes cobalt acetate, a relatively inexpensive first-row transition metal, as the primary catalyst for the key C-H activation step, avoiding the high costs associated with palladium or iridium catalysts often found in similar cross-coupling reactions. Additionally, the use of PEG-400 as a recyclable solvent system minimizes waste disposal costs and reduces the volume of organic solvents required for the reaction, leading to substantial cost savings in large-scale production environments. The high yields reported in the examples, such as the 85% yield for the iodination step and 63% for the final cyclization of Ber-2, indicate a material-efficient process that maximizes the output per kilogram of input, further driving down the unit cost of the final active pharmaceutical ingredient.
- Enhanced Supply Chain Reliability: By synthesizing the core scaffold from petrochemical-derived starting materials rather than relying on the seasonal harvest of medicinal plants, manufacturers can guarantee a consistent supply of high-quality intermediates throughout the year. The modular nature of the synthesis allows for the parallel production of different analogs using the same equipment train, providing flexibility to respond to changing clinical demands without the need for major capital investment. This reliability is crucial for maintaining the continuity of drug development programs, especially when transitioning from milligram-scale discovery to gram-scale preclinical batches.
- Scalability and Environmental Compliance: The reaction conditions, primarily involving heating in sealed tubes or round-bottom flasks at temperatures up to 140 °C, are easily scalable using standard stainless steel reactors found in most multipurpose chemical plants. The absence of extreme cryogenic conditions (below -40 °C) or high-pressure hydrogenation steps simplifies the engineering requirements for scale-up. Moreover, the workup procedures involve standard aqueous extractions and silica chromatography, which are well-understood unit operations that facilitate compliance with environmental regulations regarding solvent emissions and heavy metal waste, ensuring a greener manufacturing footprint.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these novel berberine derivatives. The answers are derived directly from the experimental data and technical specifications provided in the patent documentation, ensuring accuracy for decision-makers evaluating this technology for their pipeline. Understanding these details helps in assessing the feasibility of integrating these intermediates into existing drug discovery workflows.
Q: What is the structural advantage of the high-B-ring berberine derivatives?
A: The expansion of the B-ring to a seven-membered structure containing heteroatoms like oxygen, nitrogen, or sulfur significantly enhances the molecular flexibility and binding affinity to the IDO1/TDO enzyme active sites compared to traditional planar berberine scaffolds.
Q: Are the starting materials for this synthesis commercially available?
A: Yes, the synthesis utilizes readily available commodity chemicals such as 2,3-dimethoxybenzylamine, 3,4-methylenedioxybromobenzene, and various substituted phenylacetylenes, ensuring a robust and cost-effective supply chain for large-scale production.
Q: What is the inhibitory potency of the synthesized compounds?
A: Experimental data indicates that specific derivatives, such as Ber-1 and Ber-7, exhibit sub-micromolar IC50 values against IDO1 and TDO enzymes, demonstrating superior potency suitable for preclinical oncology research applications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Berberine Derivatives Supplier
The development of next-generation immunotherapies requires partners who can navigate the complexities of heterocyclic chemistry with precision and scale. NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop discovery to clinical supply is seamless. Our facilities are equipped with state-of-the-art rigorous QC labs capable of characterizing complex alkaloid structures and verifying stringent purity specifications, including residual solvent and heavy metal analysis, which are critical for regulatory filings. We understand that the consistency of the IDO1/TDO inhibitor supply is vital for your preclinical and clinical timelines, and we are committed to delivering batches that meet the highest international quality standards.
We invite you to engage with our technical procurement team to discuss how our manufacturing capabilities can support your specific project needs. Whether you require a Customized Cost-Saving Analysis for a multi-kilogram campaign or need to review specific COA data and route feasibility assessments for a novel analog, our experts are ready to provide the data-driven insights necessary to move your program forward. Contact us today to explore how our advanced C-H activation technologies can accelerate your oncology drug development pipeline.