Scalable Synthesis of C-Glycoside Impurities for Advanced Diabetes Drug QC
Scalable Synthesis of C-Glycoside Impurities for Advanced Diabetes Drug QC
The pharmaceutical industry's relentless pursuit of safer and more effective treatments for type 2 diabetes has placed SGLT-2 inhibitors at the forefront of therapeutic innovation. However, the regulatory landscape demands uncompromising standards for drug purity, necessitating the precise identification and quantification of even trace impurities. Patent CN113735921B introduces a groundbreaking methodology for the scalable synthesis of specific C-glycoside derivatives, addressing a critical gap in the quality control infrastructure for this class of antidiabetic agents. This technical disclosure provides a robust pathway to generate reference standards that were previously difficult to obtain in sufficient quantities, thereby empowering pharmaceutical manufacturers to meet stringent global compliance requirements with greater efficiency and confidence.
For R&D directors and quality assurance teams, the ability to access well-characterized impurity standards is not merely a regulatory checkbox but a fundamental component of risk mitigation. The method detailed in this patent elucidates a controlled chemical environment where specific structural analogues, often formed as unwanted byproducts during the main API synthesis, can be deliberately manufactured. By shifting from passive isolation to active synthesis, the industry gains a powerful tool for validating analytical methods, ensuring batch-to-batch consistency, and ultimately safeguarding patient health against potential toxicological risks associated with unknown degradants.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the acquisition of impurity reference standards for complex C-glycoside structures has been plagued by significant technical and economic hurdles. Traditional approaches typically rely on the isolation of trace impurities from large-scale production batches of the active pharmaceutical ingredient. This process is inherently inefficient, often yielding milligram quantities after extensive and costly purification efforts such as preparative HPLC. Furthermore, the structural integrity of isolated impurities can be compromised during harsh separation conditions, and the lack of a reproducible synthesis route means that supply is entirely dependent on the variability of the main manufacturing process. This unpredictability creates severe bottlenecks for supply chain managers who struggle to secure consistent materials for long-term stability studies and regulatory filings.
The Novel Approach
The methodology presented in the patent data offers a paradigm shift by establishing a dedicated, multi-step synthetic route designed specifically for the target impurity. Instead of scavenging for trace amounts, this approach constructs the molecule from defined precursors using controlled reaction conditions. The process leverages a strategic acetylation followed by a Lewis acid-catalyzed coupling and a final deprotection step. This deliberate construction allows for the optimization of each stage independently, resulting in significantly improved yields and purity profiles compared to isolation techniques. For procurement professionals, this translates to a more predictable supply chain where reference materials can be produced on demand, decoupling QC operations from the vagaries of API manufacturing campaigns.
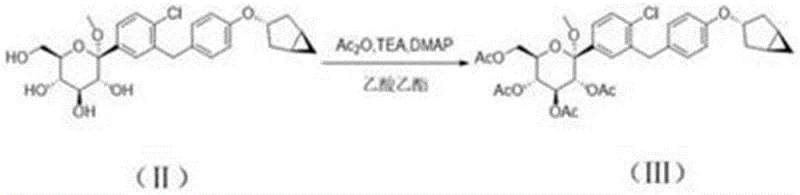
The first critical stage of this novel synthesis involves the peracetylation of the starting C-glycoside derivative. As illustrated in the reaction scheme, the substrate is treated with acetic anhydride in the presence of triethylamine and a catalytic amount of DMAP. This transformation protects the hydroxyl groups, preventing unwanted side reactions in subsequent steps and activating the anomeric center for nucleophilic attack. The use of mild conditions, specifically maintaining temperatures between 0°C and 30°C, ensures high selectivity and minimizes degradation, setting a solid foundation for the overall process efficiency.
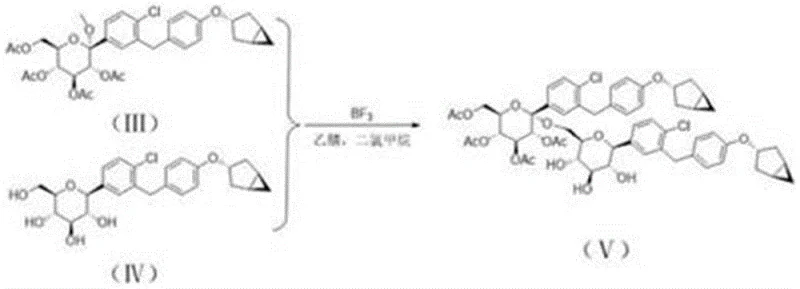
Following acetylation, the core carbon-carbon bond formation occurs through a sophisticated coupling reaction. The peracetylated intermediate reacts with a specific aglycone precursor under the influence of boron trifluoride etherate. This Lewis acid catalysis is pivotal, facilitating the formation of the C-glycosidic linkage with high stereocontrol. The reaction is conducted in acetonitrile, a solvent choice that balances solubility with reactivity, allowing the process to proceed smoothly at ambient temperatures after an initial cooling phase. This step effectively mimics the side-reaction pathway that generates the impurity in the main API synthesis but does so in a controlled manner to maximize the output of the desired reference standard.
Mechanistic Insights into Lewis Acid Catalyzed Glycosylation
Understanding the mechanistic underpinnings of this synthesis is crucial for R&D teams aiming to replicate or adapt the process for similar molecular scaffolds. The formation of the target impurity, as depicted in the overall pathway, is driven by the electrophilic activation of the anomeric center. In the presence of a strong Lewis acid like boron trifluoride, the acetoxy group at the anomeric position becomes a superior leaving group, generating an oxocarbenium ion intermediate. This highly reactive species is then intercepted by the electron-rich aromatic ring of the aglycone partner. The regioselectivity of this attack is governed by the electronic properties of the aromatic ring and the steric environment of the sugar moiety, ensuring that the carbon-carbon bond forms at the correct position to match the natural impurity profile.
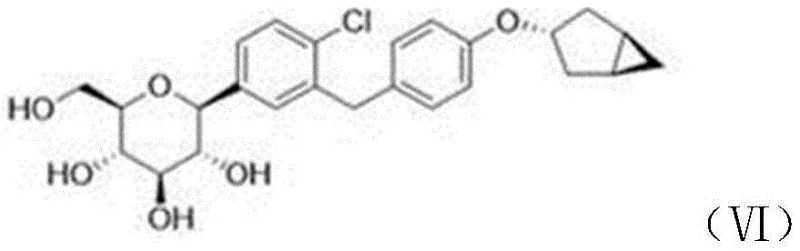
Furthermore, the stereochemical outcome of the reaction is a critical quality attribute that this method addresses with precision. The configuration at the anomeric center is preserved or controlled through the neighboring group participation of the adjacent acetyl groups, which direct the incoming nucleophile to the desired face of the sugar ring. This mechanistic feature is vital for ensuring that the synthesized impurity is structurally identical to the one found in the drug product, a requirement for valid analytical comparison. By mastering these mechanistic details, manufacturers can fine-tune reaction parameters such as temperature and stoichiometry to suppress other potential byproducts, thereby enhancing the overall purity of the final reference standard.
How to Synthesize C-Glycoside Impurity Efficiently
Implementing this synthesis protocol requires careful attention to reaction parameters and workup procedures to ensure optimal results. The process is designed to be robust, utilizing commercially available reagents and standard laboratory equipment, which facilitates easy technology transfer from R&D to pilot plant scales. The following guide outlines the critical operational phases, emphasizing the importance of temperature control and purification strategies to achieve the high purity levels necessary for regulatory reference standards. Detailed standardized synthesis steps are provided in the guide below to assist technical teams in executing this protocol effectively.
- Acetylate the starting C-glycoside derivative using acetic anhydride, TEA, and DMAP in ethyl acetate to form the peracetylated intermediate.
- Perform a Lewis acid catalyzed coupling reaction between the acetylated intermediate and the aglycone precursor using boron trifluoride etherate in acetonitrile.
- Execute a deprotection step using lithium hydroxide monohydrate in a methanol and tetrahydrofuran mixture to yield the final target impurity.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this dedicated synthesis route offers substantial strategic benefits for pharmaceutical organizations managing complex supply chains. The primary advantage lies in the decoupling of impurity standard production from the main API manufacturing schedule. Previously, delays in API production could stall QC activities due to a lack of reference materials. With a standalone synthesis, procurement managers can secure a steady inventory of critical standards, ensuring that quality control laboratories remain operational regardless of API production status. This independence significantly reduces the risk of project delays and regulatory submission hold-ups, providing a more resilient operational framework.
- Cost Reduction in Manufacturing: The elimination of preparative HPLC isolation steps represents a major driver for cost optimization. Isolation processes are notoriously resource-intensive, requiring large volumes of solvents and specialized equipment for minimal yield. By switching to a constructive synthetic approach, the consumption of expensive solvents and stationary phases is drastically reduced. Additionally, the use of common reagents like acetic anhydride and lithium hydroxide keeps raw material costs low, while the improved yields mean less starting material is wasted, leading to substantial overall cost savings in the production of these high-value reference compounds.
- Enhanced Supply Chain Reliability: The reliance on readily available, commodity-grade chemicals enhances the robustness of the supply chain. Unlike specialized catalysts or bespoke reagents that may have long lead times or single-source dependencies, the inputs for this synthesis are globally sourced and stable. This accessibility mitigates the risk of supply disruptions, ensuring that the production of impurity standards can continue uninterrupted. For supply chain heads, this reliability translates to better inventory planning and the ability to respond quickly to increased demand for QC materials during peak regulatory filing periods.
- Scalability and Environmental Compliance: The process design inherently supports scalability, moving seamlessly from gram-scale laboratory synthesis to kilogram-scale production without fundamental changes to the chemistry. The workup procedures involve standard aqueous extractions and crystallizations, which are easily adapted for large-scale reactors. Moreover, the avoidance of heavy metal catalysts simplifies waste treatment and disposal, aligning with increasingly strict environmental regulations. This green chemistry aspect not only reduces disposal costs but also enhances the corporate sustainability profile of the manufacturing operation.
Frequently Asked Questions (FAQ)
The following questions address common technical and operational inquiries regarding the implementation of this synthesis method. These insights are derived directly from the patent specifications and are intended to clarify the practical aspects of producing C-glycoside impurities. Understanding these details is essential for technical teams evaluating the feasibility of adopting this route for their specific quality control needs.
Q: Why is synthesizing specific C-glycoside impurities critical for SGLT-2 inhibitor development?
A: Regulatory agencies require rigorous identification and quantification of all impurities above specific thresholds to ensure patient safety. Synthetic reference standards allow for accurate HPLC method validation and stability testing, which is impossible with isolated trace impurities.
Q: How does this Lewis acid catalyzed route improve upon traditional isolation methods?
A: Traditional isolation from reaction mixtures yields negligible quantities with poor purity. This dedicated synthetic route allows for gram-to-kilogram scale production with controlled stereochemistry and high purity, ensuring a reliable supply for QC laboratories.
Q: What are the key scalability factors in this synthesis protocol?
A: The process utilizes common solvents like ethyl acetate and acetonitrile, avoids expensive transition metal catalysts, and employs straightforward aqueous workups. These factors significantly simplify industrial scale-up and waste management compared to complex enzymatic or multi-step catalytic routes.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable C-Glycoside Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality impurity standards play in the development and commercialization of life-saving medications. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that even the most complex synthetic challenges are met with precision and efficiency. We are committed to delivering products that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our capability to replicate and optimize patented synthesis routes allows us to provide reliable solutions for your most demanding pharmaceutical intermediate needs.
We invite you to collaborate with us to optimize your supply chain for C-glycoside derivatives and related impurities. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. By partnering with us, you gain access to specific COA data and comprehensive route feasibility assessments that can accelerate your project timelines. Contact us today to discuss how our advanced manufacturing capabilities can support your regulatory goals and ensure the continuous supply of high-purity materials essential for your success.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →