Advanced Manufacturing of 2'-Deoxy-beta-L-Nucleosides for Antiviral Drug Development
The pharmaceutical landscape for treating chronic viral infections, particularly Hepatitis B Virus (HBV), has increasingly relied on the development of L-enantiomeric nucleoside analogs due to their superior safety profiles and antiviral potency. Patent CN101415719A presents a groundbreaking methodology for the synthesis of 2'-deoxy-beta-L-nucleosides, specifically focusing on the efficient production of L-deoxycytidine (L-dC) and its valuable prodrug derivatives. This intellectual property addresses critical bottlenecks in the manufacturing of antiviral agents by introducing a robust route that converts uridine precursors into cytidine analogs with high stereoselectivity and yield. For stakeholders in the antiviral drug development sector, this technology represents a significant leap forward in process chemistry, offering a pathway to produce high-purity intermediates that are essential for next-generation therapeutics. The patent details a comprehensive strategy that encompasses the stabilization of activated sugar intermediates, the optimization of coupling reactions, and the implementation of mild amination protocols that preserve sensitive functional groups.
Historically, the conversion of uridine derivatives to cytidine analogs has been plagued by harsh reaction conditions and inefficient purification steps that hindered large-scale production. Conventional methods often relied on reagents such as Lawesson's reagent or phosphorus pentasulfide to generate 4-thio intermediates, which were subsequently aminated. These traditional approaches suffered from significant drawbacks, including the generation of toxic byproducts, unpleasant odors, and the requirement for rigorous anhydrous conditions that increased operational costs. Furthermore, the separation of the desired beta-anomer from the alpha-anomer typically necessitated expensive and time-consuming column chromatography, which is impractical for kilogram or ton-scale manufacturing. The instability of certain intermediates under acidic or basic conditions also led to racemization or decomposition, resulting in suboptimal yields and compromised product quality that failed to meet stringent pharmaceutical standards.
In stark contrast, the novel approach disclosed in the patent utilizes a sulfonyl halide activation strategy followed by direct amination, fundamentally transforming the efficiency of the synthesis. By employing reagents like tosyl chloride in the presence of phase transfer catalysts, the process activates the uracil moiety under remarkably mild conditions, avoiding the degradation pathways common in older methods. This innovation allows for the direct conversion of protected L-deoxyuridine into L-deoxycytidine derivatives with exceptional regioselectivity. The method is particularly advantageous for synthesizing amino acid ester prodrugs, such as valyl esters, as it tolerates protecting groups like Boc without causing racemization. This capability eliminates the need for complex protection-deprotection sequences, streamlining the overall synthetic route and significantly enhancing the throughput of the manufacturing process.
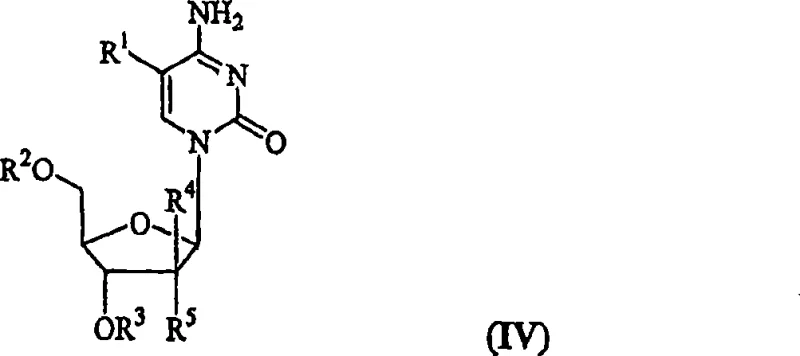
The mechanistic insights into this sulfonyl halide-catalyzed transformation reveal a sophisticated interplay between activation kinetics and thermodynamic stability. The process begins with the activation of the C-4 position of the uracil ring using a sulfonyl halide, which creates a highly reactive intermediate susceptible to nucleophilic attack. The presence of pyridinium salts as phase transfer catalysts facilitates the interaction between the organic substrate and the aminating agent, ensuring uniform reaction progress throughout the mixture. Subsequent treatment with liquid or gaseous ammonia effects the displacement of the sulfonyl group, installing the exocyclic amine required for cytosine formation. Crucially, the reaction conditions are tuned to favor the formation of the cytidine product over the starting uridine, achieving ratios as high as 12:1 or 15:1, which drastically reduces the burden on downstream purification systems.
Impurity control is another cornerstone of this technological advancement, addressing a primary concern for R&D directors focused on regulatory compliance. The patent describes a selective extraction protocol that leverages the basicity of the cytidine derivative to separate it from unreacted uridine byproducts. By forming water-soluble salts with hydrochloric acid, the cytidine species can be selectively extracted into the aqueous phase, leaving neutral uridine impurities in the organic layer. This non-chromatographic purification step is a game-changer for industrial scalability, as it replaces costly resin-based separations with simple liquid-liquid extraction units. Additionally, the use of stable halo-sugar intermediates ensures that the chiral integrity of the L-sugar is maintained throughout the coupling process, minimizing the formation of diastereomeric impurities that are difficult to remove. The result is a final product with high optical purity, meeting the rigorous specifications required for clinical-grade antiviral medications.
How to Synthesize 2'-Deoxy-beta-L-Cytidine Efficiently
The synthesis of 2'-deoxy-beta-L-cytidine and its prodrugs involves a sequence of carefully controlled chemical transformations designed to maximize yield and purity while minimizing operational complexity. The process initiates with the preparation of stable activated sugar intermediates, which serve as the foundation for high-fidelity nucleoside coupling. Following the coupling of the sugar with the base, the critical uridine-to-cytidine conversion is executed using the sulfonyl halide activation method described previously. This is followed by selective protection strategies, such as the use of Boc groups, to enable the introduction of amino acid moieties at the 3' or 5' positions. The detailed standardized synthesis steps见下方的指南 outline the specific reagents, temperatures, and workup procedures necessary to replicate this high-efficiency route in a GMP-compliant environment.
- Activate the uracil moiety of protected L-deoxyuridine using a sulfonyl halide like tosyl chloride in the presence of a phase transfer catalyst.
- Perform regioselective amination using liquid or gaseous ammonia under controlled temperature conditions to form the cytosine base.
- Purify the resulting cytidine derivative via selective extraction to remove uridine byproducts, followed by deprotection to yield the final active pharmaceutical ingredient.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis route offers substantial strategic benefits that extend beyond mere technical feasibility. The elimination of column chromatography represents a massive reduction in processing time and consumable costs, directly impacting the bottom line of manufacturing operations. By shifting to a crystallization and extraction-based purification workflow, facilities can significantly increase their batch throughput without requiring additional capital investment in separation equipment. This process intensification allows for faster turnaround times from raw material intake to finished goods, enhancing the agility of the supply chain in response to market demands for antiviral therapies. Furthermore, the use of commercially available and stable reagents like tosyl chloride reduces the risk of supply disruptions associated with specialized or hazardous chemicals.
- Cost Reduction in Manufacturing: The transition away from hazardous reagents like Lawesson's reagent and phosphorus pentasulfide leads to significant cost savings in waste disposal and safety management. Traditional methods generate sulfur-containing waste streams that require specialized treatment, whereas the new process produces benign byproducts that are easier and cheaper to handle. Additionally, the high yield of the coupling and amination steps means that less raw material is wasted, improving the overall atom economy of the synthesis. The ability to isolate intermediates as stable crystals further reduces losses during storage and transport, ensuring that the value of the material is preserved throughout the production lifecycle.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route contributes to a more resilient supply chain by reducing the dependency on fragile reaction conditions. The intermediates generated, such as the activated halo-sugars and protected nucleosides, are stable enough to be stored and shipped, allowing for flexible production scheduling and inventory management. This stability mitigates the risk of batch failures due to reagent degradation or environmental fluctuations, ensuring a consistent supply of high-quality intermediates. For global pharmaceutical companies, this reliability is crucial for maintaining continuous drug production and meeting regulatory commitments without interruption.
- Scalability and Environmental Compliance: Scaling this process from laboratory to commercial production is facilitated by the use of standard unit operations like filtration, distillation, and extraction, which are well-understood in the chemical industry. The mild reaction conditions reduce the energy consumption associated with heating and cooling, aligning with sustainability goals and reducing the carbon footprint of manufacturing. Moreover, the avoidance of heavy metals and toxic sulfur reagents simplifies environmental compliance, making it easier to obtain permits and operate in regions with strict ecological regulations. This environmental compatibility enhances the long-term viability of the manufacturing site and reduces the risk of regulatory penalties.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this L-nucleoside synthesis technology. These answers are derived directly from the experimental data and embodiments provided in the patent documentation, ensuring accuracy and relevance for potential partners. Understanding these details is essential for evaluating the feasibility of integrating this process into existing manufacturing pipelines or for initiating new development projects focused on antiviral drug candidates.
Q: What are the advantages of using sulfonyl halides over Lawesson's reagent for cytidine synthesis?
A: Sulfonyl halides like tosyl chloride offer milder reaction conditions and avoid the toxicity and odor associated with phosphorus-sulfur reagents, leading to safer industrial scale-up and easier waste management.
Q: How does this process ensure high stereochemical purity of L-nucleosides?
A: The process utilizes stable activated halo-sugar intermediates that maintain chiral integrity during coupling, combined with selective crystallization steps that effectively separate alpha and beta anomers.
Q: Can this method be applied to the synthesis of amino acid ester prodrugs?
A: Yes, the methodology is specifically designed to tolerate amino acid protecting groups like Boc, allowing for the direct synthesis of 3'-O-aminoacyl prodrugs such as valyl esters without racemization.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2'-Deoxy-beta-L-Cytidine Supplier
As the demand for effective antiviral therapies continues to grow, the ability to manufacture high-quality L-nucleoside intermediates at scale becomes a critical competitive advantage. NINGBO INNO PHARMCHEM stands at the forefront of this field, leveraging advanced synthetic methodologies like those described in CN101415719A to deliver superior products to the global market. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the volume requirements of major pharmaceutical clients. We are committed to maintaining stringent purity specifications through our rigorous QC labs, guaranteeing that every batch of 2'-deoxy-beta-L-cytidine or its prodrugs meets the highest industry standards for safety and efficacy.
We invite you to collaborate with us to explore the full potential of this innovative synthesis route for your drug development programs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific production needs, demonstrating how this technology can optimize your budget. Please contact us to request specific COA data and route feasibility assessments, and let us partner with you to accelerate the delivery of life-saving antiviral medications to patients worldwide.