Advanced Synthesis of Antiviral Protease Inhibitors Using Carbohydrate Precursors for Commercial Scale-up
The pharmaceutical industry continuously seeks more efficient pathways for producing complex active pharmaceutical ingredients (APIs), particularly in the critical area of antiviral therapeutics. Patent CN1118476C introduces a groundbreaking approach to synthesizing aspartyl protease inhibitors, specifically targeting Human Immunodeficiency Virus (HIV) treatments. This technology pivots away from the laborious construction of linear peptidomimetics, instead leveraging the inherent chirality and structural rigidity of carbohydrate precursors. By utilizing L-mannose-1,4:6,3-dilactone as a central scaffold, the patent outlines a facile two-step synthesis that dramatically streamlines the production of compounds with nanomolar antiviral activity. For R&D directors and procurement specialists, this represents a significant opportunity to optimize supply chains for high-purity pharmaceutical intermediates while reducing the environmental footprint associated with multi-step organic synthesis.
The strategic value of this patent lies in its ability to bypass the synthetic bottlenecks that have historically plagued the manufacturing of protease inhibitors. Traditional methods often involve protecting group manipulations that add cost and time without adding value to the final molecular architecture. In contrast, the methodology described in CN1118476C capitalizes on the natural symmetry and functional group distribution of the mannose backbone. This allows for the simultaneous installation of P1 and P1' binding moieties through straightforward O-alkylation, followed by the introduction of P2 and P2' groups via ring-opening amidation. This logical disconnection of the target molecule not only enhances overall yield potential but also ensures consistent stereochemical integrity, a critical parameter for regulatory approval and clinical efficacy.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of HIV protease inhibitors has been characterized by extreme complexity and low overall efficiency. As noted in the background of the patent, established drugs like Saquinavir require synthetic routes spanning approximately 20 steps, resulting in dismal overall yields of around 2 percent. These conventional pathways typically rely on the assembly of linear peptide chains, necessitating extensive use of protecting groups to manage the reactivity of multiple amino and carboxyl functionalities. Each protection and deprotection cycle introduces additional unit operations, increases solvent consumption, and generates substantial chemical waste. Furthermore, the reliance on chiral pool amino acids often requires resolution steps or expensive chiral catalysts to ensure the correct stereochemistry at every center, compounding the cost and logistical challenges for supply chain managers.
Beyond the sheer number of steps, prior art methods frequently suffer from chemoselectivity issues that compromise product quality. For instance, attempts to introduce ether linkages at specific positions on a carbohydrate or peptide backbone using triflate activation have been shown to favor elimination reactions, producing unwanted alkene byproducts rather than the desired substitution. This lack of control necessitates rigorous purification protocols, such as repeated chromatography or recrystallization, which are difficult to translate from the laboratory bench to multi-ton commercial production. The cumulative effect of these inefficiencies is a high cost of goods sold (COGS) and a fragile supply chain vulnerable to raw material shortages or process deviations, making the search for alternative synthetic strategies an urgent priority for the industry.
The Novel Approach
The methodology disclosed in CN1118476C offers a transformative solution by reimagining the core scaffold of the inhibitor. Instead of building up from individual amino acids, the process starts with L-mannaric acid dilactone, a commercially available or easily accessible carbohydrate derivative. This starting material inherently possesses the necessary carbon backbone and stereochemical configuration required for the final drug substance. The novelty lies in the direct O-alkylation of the dilactone at the C-2 and C-5 positions, which effectively installs the P1 and P1' side chains in a single operational sequence. This is followed by a regioselective ring-opening reaction with amines to form the terminal amide bonds, completing the assembly of the pharmacophore with remarkable economy of motion.
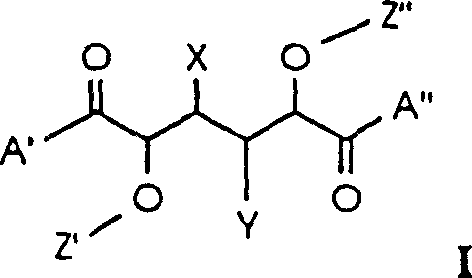
This new approach fundamentally alters the manufacturing landscape by reducing the step count and eliminating the need for problematic activation reagents. The use of stable lactone intermediates allows for better control over reaction kinetics and thermal profiles, which is essential for safe scale-up. Moreover, the flexibility of the route permits the generation of a diverse library of analogs by simply varying the alkylating agents or the amine nucleophiles, facilitating rapid structure-activity relationship (SAR) studies without redesigning the entire synthesis. For procurement teams, this translates to a more robust sourcing strategy where key intermediates can be stockpiled, and for R&D, it provides a versatile platform for developing next-generation antiviral agents with improved resistance profiles and pharmacokinetic properties.
Mechanistic Insights into Carbohydrate-Based Scaffold Assembly
The core mechanistic advantage of this synthesis revolves around the unique reactivity of the 1,4:6,3-dilactone system derived from L-mannose. Unlike open-chain sugars which exist in equilibrium between various anomeric forms, the dilactone structure is locked into a rigid conformation that pre-organizes the hydroxyl groups for selective functionalization. The initial O-alkylation step typically employs benzyl trichloroacetimidate or similar activated alkylating agents in the presence of a Lewis acid catalyst like trimethylsilyl triflate. This reaction proceeds through an oxocarbenium ion-like transition state where the C-2 and C-5 hydroxyls, being secondary and strategically positioned, act as nucleophiles to capture the electrophilic alkyl species. The result is the formation of stable ether linkages that define the P1 and P1' pockets of the enzyme inhibitor, crucial for binding affinity.
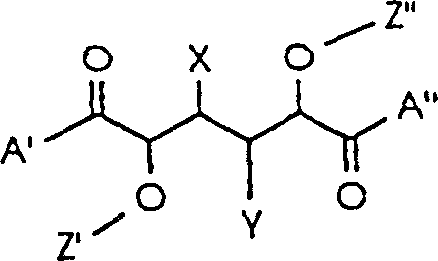
Following alkylation, the ring-opening step exploits the electrophilicity of the lactone carbonyls. When treated with primary or secondary amines, such as N-methyl-L-valine amide or amino-indanol derivatives, the lactone rings undergo nucleophilic acyl substitution. This transformation converts the cyclic esters into acyclic amides, simultaneously establishing the peptide-mimetic bonds at the termini of the molecule. The stereochemistry at the newly formed centers is preserved due to the rigidity of the intermediate, ensuring that the final product maintains the (2R, 3R, 4R, 5R) configuration essential for biological activity. This mechanistic pathway avoids the racemization risks often associated with peptide coupling reagents, thereby delivering a product with superior optical purity and reduced impurity burden.
Impurity control is further enhanced by the avoidance of elimination-prone leaving groups. In previous methodologies, converting hydroxyls to triflates often led to competing E2 elimination reactions, generating olefinic impurities that were structurally similar to the product and difficult to remove. The current process bypasses this issue entirely by using direct alkylation and amidation, which are inherently cleaner transformations. Any minor byproducts formed, such as mono-alkylated species or hydrolysis products, possess significantly different polarity profiles compared to the target molecule, allowing for efficient removal via standard crystallization or extraction techniques. This high level of chemical fidelity is critical for meeting the stringent purity specifications required for clinical-grade pharmaceutical intermediates.
How to Synthesize Antiviral Protease Inhibitors Efficiently
Implementing this synthesis route requires careful attention to reaction conditions to maximize yield and purity. The process begins with the preparation of the O-alkylated dilactone intermediate, which serves as the pivotal building block for the entire series. Operators must ensure anhydrous conditions during the alkylation step to prevent hydrolysis of the sensitive lactone rings. Subsequent ring-opening reactions should be monitored closely to prevent over-reaction or degradation of the amine components. The versatility of this method allows for the incorporation of various P2 and P2' groups, enabling the customization of the final inhibitor for specific therapeutic needs. Detailed standardized operating procedures for each stage are essential to ensure reproducibility across different manufacturing sites.
- Perform O-alkylation on L-mannose-1,4: 6,3-dilactone to introduce Z' and Z'' groups at the C-2 and C-5 positions using suitable alkylating agents.
- Open the lactone rings by reacting the dialkylated intermediate with primary or secondary amines to form the A' and A'' terminal groups.
- Optionally modify the C-3 or C-4 hydroxyl groups to convert them into X and Y substituents such as hydrogen or methoxy groups as required.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this carbohydrate-based synthesis offers profound benefits for cost management and supply chain resilience. The primary driver of cost reduction is the drastic simplification of the synthetic route. By condensing what was previously a 20-step sequence into a much shorter pathway, manufacturers can significantly lower labor costs, utility consumption, and equipment occupancy time. The elimination of multiple protection and deprotection cycles also reduces the volume of solvents and reagents required, leading to substantial savings in raw material procurement and waste disposal fees. This lean manufacturing approach directly improves the gross margin profile of the final API, making it more competitive in the global marketplace.
- Cost Reduction in Manufacturing: The streamlined process eliminates the need for expensive chiral catalysts and complex peptide coupling reagents that are typical in conventional protease inhibitor synthesis. By relying on abundant carbohydrate starting materials like L-mannose, the cost of goods is anchored to commodity pricing rather than specialized fine chemicals. Furthermore, the high atom economy of the ring-opening reaction ensures that a larger proportion of the input mass ends up in the final product, minimizing waste generation and associated disposal costs. This efficiency translates into a more sustainable and economically viable production model.
- Enhanced Supply Chain Reliability: Sourcing risks are mitigated by the use of widely available starting materials. L-mannose and its derivatives are produced on a large scale for the food and fermentation industries, ensuring a stable and diversified supply base. Unlike proprietary intermediates that may be sourced from a single vendor, these carbohydrate precursors can be procured from multiple qualified suppliers, reducing the risk of supply disruptions. Additionally, the robustness of the chemical steps allows for flexible manufacturing schedules, enabling producers to respond quickly to fluctuations in market demand without compromising product quality.
- Scalability and Environmental Compliance: The chemistry described is inherently scalable, utilizing unit operations such as crystallization and filtration that are well-suited for large-scale reactors. The avoidance of hazardous reagents like thionyl chloride or phosphorus halides simplifies safety management and reduces the regulatory burden associated with handling toxic substances. Moreover, the reduced solvent usage and waste generation align with green chemistry principles, helping companies meet increasingly strict environmental regulations and corporate sustainability goals. This compliance advantage facilitates faster regulatory approvals and smoother audits.
Frequently Asked Questions (FAQ)
Understanding the technical nuances of this synthesis is vital for stakeholders evaluating its implementation. The following questions address common inquiries regarding the process capabilities, quality attributes, and strategic fit within a broader pharmaceutical portfolio. These answers are derived directly from the technical disclosures within the patent documentation, providing a factual basis for decision-making.
Q: What is the primary advantage of using L-mannose dilactone over traditional peptide mimetics?
A: The use of L-mannose dilactone significantly reduces the number of synthetic steps compared to conventional linear peptidomimetics like Saquinavir, which often require up to 20 steps. This carbohydrate-based approach simplifies stereocontrol and utilizes readily available starting materials.
Q: How does this synthesis method impact impurity profiles?
A: The method avoids the use of triflate activation which can lead to elimination side reactions forming alkenes. Instead, it relies on direct O-alkylation and ring-opening, resulting in a cleaner reaction profile and easier purification of the final protease inhibitor intermediates.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the process is designed for scalability. It utilizes robust reaction conditions such as O-alkylation in aprotic solvents and ring-opening with amines that can be controlled for exotherms. The formation of crystalline intermediates facilitates isolation and purification on a commercial scale.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Antiviral Protease Inhibitors Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthesis routes in the development of life-saving antiviral medications. Our team of expert chemists has extensively analyzed the methodology presented in CN1118476C and possesses the technical capability to execute this carbohydrate-based strategy with precision. We offer comprehensive CDMO services tailored to the unique challenges of protease inhibitor production, ensuring that your project moves seamlessly from process development to commercial manufacturing. Our commitment to excellence guarantees that every batch meets the highest standards of quality and consistency.
We invite you to leverage our expertise to optimize your supply chain and reduce production costs. With extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, we are equipped to handle projects of any size. Our facilities feature stringent purity specifications and rigorous QC labs to ensure that all intermediates and APIs comply with global regulatory requirements. Contact our technical procurement team today to request a Customized Cost-Saving Analysis, specific COA data, and route feasibility assessments for your next antiviral development program.