Advanced Synthesis of Flumatinib Intermediate via Copper-Catalyzed Amidation for Commercial Scale-up
Advanced Synthesis of Flumatinib Intermediate via Copper-Catalyzed Amidation for Commercial Scale-up
The pharmaceutical industry's demand for high-purity oncology therapeutics continues to drive innovation in process chemistry, particularly for complex kinase inhibitors like Flumatinib. Patent CN111763170A, published in October 2020, discloses a groundbreaking preparation method for a critical Flumatinib intermediate that fundamentally restructures the synthetic landscape for this antitumor agent. This technology addresses long-standing challenges in the manufacturing of Protein Tyrosine Kinase (PTK) inhibitors by introducing a robust, copper-catalyzed coupling strategy that bypasses traditional, hazardous acyl chloride methodologies. For R&D directors and supply chain leaders, this patent represents a pivotal shift towards greener, more cost-effective, and scalable production of high-purity pharmaceutical intermediates. The core innovation lies in the convergent synthesis of N-(6-methyl-5-nitropyridin-3-yl)-4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)benzamide, a key precursor that ensures the structural integrity and safety profile of the final API.
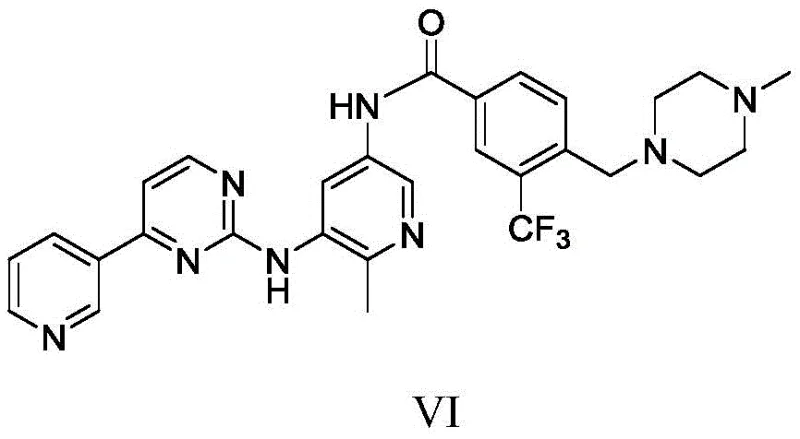
Flumatinib acts by inhibiting Bcr-Abl tyrosine kinase activity, effectively suppressing tumor cell proliferation in Philadelphia chromosome-positive Chronic Myeloid Leukemia (CML) and acute lymphocytic leukemia. The structural complexity of this molecule, featuring a trifluoromethyl-substituted benzamide linked to a nitropyridine moiety via an amine bridge, necessitates precise synthetic control to avoid deleterious impurities. The disclosed method not only streamlines the assembly of this scaffold but also aligns with modern regulatory expectations regarding genotoxic impurity (GTI) control. By leveraging this proprietary route, manufacturers can secure a reliable flumatinib intermediate supplier status, offering downstream API producers a material that is both chemically superior and environmentally compliant.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
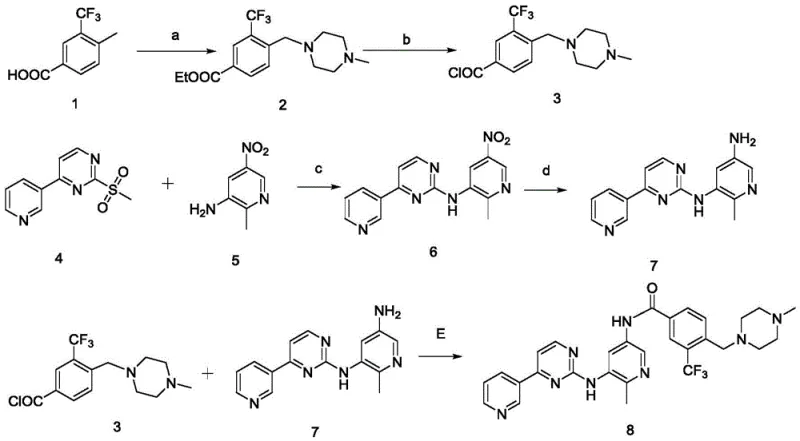
Historically, the synthesis of Flumatinib and its precursors has been plagued by significant operational and safety hurdles, primarily stemming from the reliance on acyl chloride chemistry. As illustrated in prior art literature, such as the routes reported by Xugang et al., conventional methods often involve the activation of carboxylic acids using thionyl chloride or oxalyl chloride to form reactive acyl chlorides. While chemically effective, this approach introduces severe liabilities: acyl chlorides are classified as genotoxic warning structures, posing a critical risk of contaminating the final drug substance with mutagenic impurities that are difficult to purge. Furthermore, the generation of large volumes of strong acidic wastewater during the quenching and workup phases creates a substantial environmental burden, escalating waste treatment costs and complicating regulatory compliance. Additionally, traditional routes frequently depend on intermediates like 6-methyl-5-nitropyridine-3-amine, which are notoriously difficult to source commercially, leading to supply chain bottlenecks and inflated raw material costs.
The Novel Approach
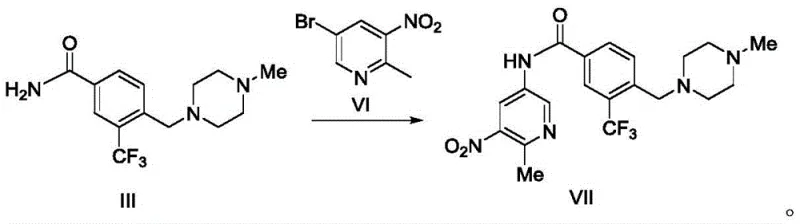
In stark contrast to these legacy methods, the novel approach detailed in CN111763170A employs a strategic disconnection that entirely circumvents the use of acyl chlorides. Instead of activating the benzoic acid derivative as a chloride, the process utilizes a direct amidation to form the benzamide fragment, followed by a sophisticated copper-catalyzed C-N bond formation to link the pyridine and benzamide domains. This methodology not only eliminates the genotoxic risk associated with acyl halides but also drastically simplifies the purification workflow. The key coupling reaction between the benzamide (Compound III) and the bromo-nitropyridine (Compound VI) proceeds under mild conditions using a copper catalyst and a diamine ligand, delivering the target intermediate in high yield. This shift represents a paradigm change in cost reduction in API manufacturing, transforming a hazardous, multi-step sequence into a streamlined, safe, and highly efficient process suitable for modern industrial standards.
Mechanistic Insights into Copper-Catalyzed C-N Coupling
The heart of this technological advancement is the copper-catalyzed amidation reaction, which facilitates the formation of the critical aryl-amine bond between the electron-deficient pyridine ring and the benzamide nitrogen. Mechanistically, this transformation likely proceeds via a Ullmann-type condensation or a related copper-mediated cross-coupling cycle. The reaction initiates with the coordination of the copper(I) species, such as cuprous bromide or cuprous iodide, to the diamine ligand (e.g., N,N'-dimethylethylenediamine or 1,2-cyclohexanediamine), forming an active catalytic complex. This complex then undergoes oxidative addition with the aryl bromide (Compound VI), followed by coordination and deprotonation of the amide nitrogen from Compound III by the inorganic base (potassium carbonate or potassium phosphate). The subsequent reductive elimination step releases the coupled product (Compound VII) and regenerates the active copper catalyst. The choice of solvent plays a pivotal role; while polar aprotic solvents like DMF or DMSO are common, the patent highlights the efficacy of toluene and 1,4-dioxane, which offer easier removal and lower toxicity profiles, further enhancing the process's green chemistry credentials.
From an impurity control perspective, this mechanism offers distinct advantages over nucleophilic aromatic substitution (SnAr) strategies that might require harsher conditions or more reactive leaving groups. The specificity of the copper catalyst minimizes side reactions such as homocoupling of the aryl halide or degradation of the sensitive nitro group. Moreover, the reaction conditions—typically maintained between 90°C and 100°C—are sufficiently energetic to drive the coupling to completion without compromising the thermal stability of the intermediates. The result is a crude reaction mixture with a remarkably clean profile, allowing for simple crystallization from ammonia water to achieve HPLC purities exceeding 99%. This high level of chemical fidelity is crucial for R&D teams focused on minimizing downstream purification loads and ensuring consistent batch-to-batch quality for regulatory filings.
How to Synthesize Flumatinib Intermediate Efficiently
The execution of this synthesis requires precise control over stoichiometry and reaction parameters to maximize yield and purity. The process begins with the preparation of the two key fragments: the benzamide derivative (III) and the bromo-nitropyridine (VI). The benzamide is synthesized via a direct ammonolysis of the corresponding carboxylic acid using sulfamide or ammonia sources, avoiding chlorination entirely. Simultaneously, the pyridine fragment is accessed through a methylation and hydrolysis sequence starting from commercially available 5-bromo-2-chloro-3-nitropyridine. Once these building blocks are secured, they are combined in the presence of the copper catalyst system. The detailed standardized synthetic steps, including specific molar ratios, temperature profiles, and workup procedures, are outlined in the guide below.
- Synthesize 4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)benzamide (III) via ammonolysis of the corresponding acid.
- Prepare 5-bromo-2-methyl-3-nitropyridine (VI) through alkylation and hydrolysis of 5-bromo-2-chloro-3-nitropyridine.
- Perform the key coupling reaction between Compound III and Compound VI using a Copper(I) catalyst, diamine ligand, and base in toluene or dioxane.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthetic route translates into tangible strategic benefits that extend beyond mere chemical elegance. The primary advantage lies in the drastic simplification of the supply chain for raw materials. By utilizing 5-bromo-2-chloro-3-nitropyridine—a commodity chemical widely available from global suppliers—the process eliminates dependence on obscure, custom-synthesized intermediates that often suffer from long lead times and price volatility. This shift ensures a more resilient supply chain, reducing the risk of production stoppages due to material shortages. Furthermore, the elimination of acyl chloride reagents removes the need for specialized corrosion-resistant equipment and extensive scrubbing systems required to handle corrosive gases like HCl and SO2, thereby lowering capital expenditure (CAPEX) and operational expenditure (OPEX) for manufacturing facilities.
- Cost Reduction in Manufacturing: The economic impact of this process is profound, driven principally by the removal of expensive and hazardous reagents. Traditional acyl chloride methods require stoichiometric amounts of chlorinating agents and generate significant quantities of salt waste, the disposal of which is increasingly costly under strict environmental regulations. By replacing this with a catalytic coupling using inexpensive copper salts and common bases like potassium carbonate, the direct material costs are significantly reduced. Additionally, the high yield (up to 96% in optimized examples) and high purity minimize the loss of valuable intermediates during purification, maximizing the overall mass efficiency of the plant. This efficiency directly contributes to a lower cost of goods sold (COGS), providing a competitive pricing advantage in the global market for oncology intermediates.
- Enhanced Supply Chain Reliability: Supply continuity is paramount in the pharmaceutical sector, where delays can impact patient access to life-saving medications. This new method enhances reliability by shortening the synthetic sequence and relying on robust, scalable chemistry. The reaction tolerances are broad, with successful outcomes reported across a range of ligands and solvents, providing flexibility in sourcing. If a specific ligand becomes unavailable, alternatives like ethylenediamine or cyclohexanediamine can be substituted without re-validating the entire process. This flexibility, combined with the use of stable, non-hygroscopic reagents, ensures that production schedules can be maintained consistently, reducing lead time for high-purity pharmaceutical intermediates and fostering stronger partnerships with API manufacturers.
- Scalability and Environmental Compliance: Scaling chemical processes from the laboratory to the tonnage scale often reveals hidden complexities, but this route is inherently designed for commercial scale-up of complex pharmaceutical intermediates. The use of toluene or dioxane as solvents facilitates easy recovery and recycling via distillation, aligning with solvent selection guides for sustainable manufacturing. Moreover, the absence of genotoxic reagents simplifies the cleaning validation process between batches, reducing downtime and cross-contamination risks. The wastewater generated is significantly less acidic and toxic compared to acyl chloride routes, easing the burden on effluent treatment plants and ensuring compliance with increasingly stringent environmental protection laws. This environmental stewardship not only mitigates regulatory risk but also enhances the corporate social responsibility profile of the manufacturing entity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented technology. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing a clear understanding of the process capabilities and limitations. Understanding these details is essential for technical teams evaluating the feasibility of technology transfer and for commercial teams negotiating supply agreements.
Q: How does this new method address genotoxic impurity concerns in Flumatinib synthesis?
A: The novel route completely avoids the use of acyl chloride intermediates, which are known genotoxic warning structures. By utilizing a direct amidation strategy followed by copper-catalyzed coupling, the process eliminates the risk of residual genotoxic reagents and simplifies impurity control.
Q: What are the primary cost drivers reduced in this synthetic pathway?
A: Cost reduction is achieved by eliminating expensive chlorinating agents and the associated waste treatment for strong acidic wastewater. Additionally, the use of readily available starting materials like 5-bromo-2-chloro-3-nitropyridine replaces hard-to-source intermediates, stabilizing raw material costs.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process is designed for scalability. It utilizes robust reaction conditions (90-100°C), common solvents like toluene, and simple crystallization for purification, achieving yields over 90% and HPLC purity exceeding 99% in optimized examples.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Flumatinib Intermediate Supplier
The transition from laboratory innovation to commercial reality requires a partner with deep technical expertise and proven manufacturing capabilities. NINGBO INNO PHARMCHEM stands at the forefront of this transition, offering comprehensive CDMO services tailored to the complex needs of oncology drug development. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the promising yields and purities demonstrated in patent CN111763170A can be reliably replicated on an industrial scale. We adhere to stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of Flumatinib intermediate meets the exacting standards required for GMP API synthesis.
We invite global pharmaceutical partners to collaborate with us to leverage this advanced synthetic route for their Flumatinib programs. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis that quantifies the economic benefits of switching to this greener, more efficient methodology. We encourage potential clients to contact us directly to obtain specific COA data from our pilot batches and to discuss detailed route feasibility assessments. Together, we can accelerate the delivery of high-quality, affordable cancer therapies to patients worldwide while optimizing your supply chain for the future.