Advanced Synthesis of 6-(Dibromomethyl)-2-methylquinazoline-4(3H)-one for Commercial API Production
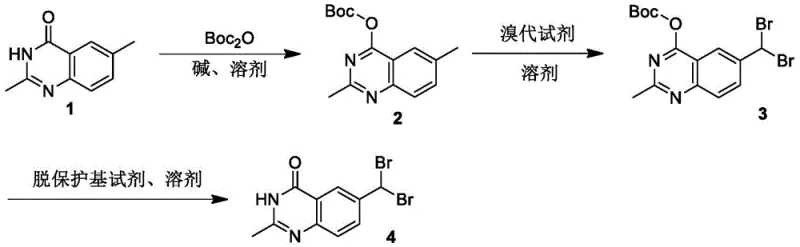
The pharmaceutical industry continuously seeks robust synthetic routes for critical oncology intermediates, particularly for established therapies like Raltitrexed. Patent CN111995588B discloses a groundbreaking method for synthesizing 6-(dibromomethyl)-2-methylquinazoline-4(3H)-one, a pivotal compound serving both as a key medical intermediate and an essential reference standard for impurity profiling. Raltitrexed, a potent thymidylate synthase inhibitor, remains a first-line treatment for advanced colorectal cancer, necessitating rigorous quality control measures where this specific dibromomethyl derivative plays an irreplaceable role. Historically, the lack of reported synthetic routes for this specific impurity standard created bottlenecks in analytical validation and supply chain stability for generic manufacturers. This new methodology addresses these challenges by introducing a strategic protection-deprotection sequence that fundamentally alters the reactivity profile of the quinazoline core, enabling unprecedented selectivity.
For research and development directors, the significance of this patent lies in its ability to generate high-purity reference materials that were previously difficult to access. The structural integrity of the quinazoline ring is preserved while functionalizing the methyl group with high precision. This level of control is paramount when synthesizing standards used to validate the safety and efficacy of active pharmaceutical ingredients (APIs). By establishing a reliable pathway to this complex heterocycle, the patent opens new avenues for derivative synthesis, allowing medicinal chemists to explore novel small molecule drugs based on the quinazoline scaffold with greater confidence in their starting materials.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional approaches to functionalizing methyl groups on heterocyclic rings like quinazoline often suffer from poor regioselectivity, particularly when multiple reactive sites are present. In the case of 2,6-dimethylquinazoline-4(3H)-one, direct bromination attempts typically result in a chaotic mixture of products. Without specific directing groups or protection strategies, radical bromination agents cannot easily distinguish between the methyl group at the 2-position and the methyl group at the 6-position. This lack of discrimination leads to the formation of mono-brominated, di-brominated, and mixed-isomer byproducts that are notoriously difficult to separate. The resulting crude mixtures require extensive and costly purification processes, such as repeated recrystallization or preparative chromatography, which drastically reduce overall throughput and increase the cost of goods sold.
Furthermore, conventional methods often struggle with the solubility and polarity characteristics of the starting quinazoline ketones. The inherent polarity of the unprotected NH group in the quinazoline ring can lead to aggregation or poor solubility in non-polar organic solvents commonly used for radical reactions. This physical limitation restricts the choice of reaction media and can lead to incomplete conversions or heterogeneous reaction conditions that are difficult to control on a large scale. For procurement managers, these inefficiencies translate into volatile pricing and unreliable supply timelines, as manufacturers struggle to consistently meet purity specifications required by regulatory bodies for oncology drugs.
The Novel Approach
The innovative route described in the patent overcomes these historical hurdles by employing a temporary Boc (tert-butoxycarbonyl) protecting group on the ring nitrogen. This strategic modification effectively masks the polar NH functionality, significantly reducing the overall polarity of the molecule and enhancing its solubility in standard organic solvents like dichloromethane and chloroform. More importantly, the introduction of the bulky Boc group exerts a profound steric and electronic influence on the quinazoline system. It electronically deactivates certain positions while sterically shielding the 2-methyl group, thereby channeling the reactivity of the brominating agent exclusively toward the 6-methyl position. This results in a highly selective transformation that yields the desired 6-(dibromomethyl) product with minimal formation of the unwanted 2-brominated isomer.
Following the selective bromination, the protecting group is cleanly removed under mild acidic conditions, regenerating the parent quinazoline structure with the desired functionalization intact. This three-step sequence—protection, selective functionalization, and deprotection—represents a paradigm shift in how complex heterocyclic intermediates are manufactured. It transforms a potentially low-yielding, messy reaction into a streamlined, high-fidelity process. For supply chain heads, this reliability is invaluable, as it ensures consistent batch-to-batch quality and reduces the risk of production delays caused by failed purification steps or out-of-specification impurity profiles.
Mechanistic Insights into Boc-Mediated Selective Bromination
The core mechanistic advantage of this synthesis lies in the modulation of the quinazoline ring's electronic density through N-protection. In the unprotected state, the electron-withdrawing nature of the carbonyl and the ring nitrogens creates a complex electronic environment where radical abstraction of hydrogen atoms from either methyl group is competitive. However, upon conversion to the tert-butyl (2,6-dimethylquinazolin-4-yl) carboxylate (Compound 2), the electronic landscape shifts. The Boc group, while electron-withdrawing by induction, alters the resonance contributions of the ring nitrogen, effectively changing the bond dissociation energies of the adjacent C-H bonds. The steric bulk of the tert-butyl moiety physically obstructs the approach of the radical species to the 2-methyl group, which is spatially closer to the protected nitrogen than the 6-methyl group.
Consequently, when N-bromosuccinimide (NBS) is introduced in the presence of a radical initiator like benzoyl peroxide (BPO), the bromine radicals selectively abstract hydrogen atoms from the more accessible and electronically favorable 6-methyl group. This generates a stable radical intermediate at the 6-position, which subsequently reacts with another equivalent of NBS to form the dibromomethyl functionality. The reaction proceeds with high fidelity because the activation energy for bromination at the shielded 2-position is significantly higher. This mechanistic understanding allows for precise control over reaction stoichiometry; using approximately 2 equivalents of NBS ensures complete conversion to the dibromo species without excessive over-bromination of the aromatic ring itself. The final acidic deprotection step utilizes the acid-lability of the Boc group, where protonation of the carbamate oxygen facilitates the elimination of isobutylene and carbon dioxide, cleanly restoring the NH functionality of the quinazolinone.
Impurity control is inherently built into this mechanism. By preventing the formation of the 2-brominated isomer at the source, the process eliminates the most difficult-to-remove impurity from the crude stream. Any minor side products, such as mono-brominated intermediates, are easily separated during the workup or final crystallization due to differences in polarity and solubility compared to the target dibromide. This intrinsic purity is critical for the compound's application as a reference standard, where even trace levels of isomeric impurities can skew analytical results for the final drug product. The use of mild acidic reagents like trifluoroacetic acid (TFA) or hydrochloric acid for deprotection further ensures that the sensitive dibromomethyl group remains stable, avoiding hydrolysis to the aldehyde or other degradation pathways that might occur under harsher conditions.
How to Synthesize 6-(Dibromomethyl)-2-methylquinazoline-4(3H)-one Efficiently
The synthesis protocol outlined in the patent provides a clear, scalable roadmap for producing this high-value intermediate. The process begins with the protection of 2,6-dimethylquinazoline-4(3H)-one using di-tert-butyl dicarbonate (Boc2O) and a suitable base such as sodium hydride or potassium carbonate in anhydrous THF or DMF. This step requires careful temperature control, typically initiating at low temperatures to manage exotherms before warming to facilitate completion. The second stage involves the crucial radical bromination, where the protected intermediate is treated with NBS and a catalytic amount of BPO in a halogenated solvent like chloroform or dichloromethane under reflux conditions. The final step is a straightforward acidic workup where the Boc group is cleaved, followed by neutralization and crystallization to isolate the pure product. Detailed standardized operating procedures for each unit operation are essential for maintaining consistency.
- Protect the quinazoline nitrogen with a Boc group using base and Boc2O to modulate reactivity.
- Perform selective radical bromination on the 6-methyl group using NBS and a radical initiator.
- Remove the Boc protecting group under acidic conditions to yield the final dibromomethyl product.
Commercial Advantages for Procurement and Supply Chain Teams
Adopting this novel synthetic route offers substantial strategic benefits for organizations managing the supply of oncology intermediates. The primary advantage is the drastic simplification of the purification train. By achieving high regioselectivity through chemical design rather than relying on downstream separation technologies, manufacturers can significantly reduce solvent consumption and waste generation. This aligns perfectly with modern green chemistry initiatives and reduces the environmental footprint of the manufacturing process. For procurement teams, this efficiency translates into a more stable cost structure, as the yield losses associated with discarding off-spec isomers are virtually eliminated. The reliance on commodity chemicals like NBS, Boc2O, and common organic solvents further insulates the supply chain from the volatility associated with specialized catalysts or rare reagents.
- Cost Reduction in Manufacturing: The elimination of complex chromatographic separations required to remove 2-position isomers leads to significant operational savings. Traditional methods often incur high costs due to low yields and expensive purification media; this new route bypasses those bottlenecks entirely. The high selectivity ensures that the majority of the starting material is converted directly into the desired product, maximizing atom economy and reducing the cost per kilogram of the final API intermediate. Furthermore, the mild reaction conditions reduce energy consumption associated with heating and cooling, contributing to lower utility costs across the production lifecycle.
- Enhanced Supply Chain Reliability: The robustness of this three-step sequence ensures consistent delivery timelines. Because the chemistry is less prone to failure modes associated with mixed product formation, batch failure rates are minimized. This reliability is critical for maintaining the continuity of supply for Raltitrexed production, where interruptions can have severe clinical implications. The use of stable, shelf-stable reagents means that raw material inventory can be managed more effectively, reducing the risk of production stoppages due to reagent degradation or scarcity. This predictability allows supply chain planners to optimize inventory levels and reduce safety stock requirements.
- Scalability and Environmental Compliance: The process is inherently scalable, utilizing unit operations such as filtration, extraction, and crystallization that are standard in multi-ton manufacturing facilities. The solvents employed, primarily dichloromethane and ethyl acetate, are well-understood in terms of recovery and recycling, facilitating compliance with strict environmental regulations regarding volatile organic compound (VOC) emissions. The absence of heavy metal catalysts removes the need for expensive and time-consuming metal scavenging steps, simplifying the regulatory filing process for the final drug substance. This ease of scale-up supports the commercial scale-up of complex heterocycles without the need for specialized reactor configurations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of this quinazoline derivative. These insights are derived directly from the experimental data and technical disclosures within the patent documentation, providing clarity for stakeholders evaluating this technology for integration into their supply chains. Understanding these nuances is key to leveraging the full potential of this synthetic advancement.
Q: Why is 6-(dibromomethyl)-2-methylquinazoline-4(3H)-one critical for Raltitrexed production?
A: This compound serves as an irreplaceable reference substance for detecting related substances and impurities in Raltitrexed bulk drugs, ensuring strict quality control compliance for this first-line colorectal cancer treatment.
Q: How does the Boc-protection strategy improve synthesis selectivity?
A: By introducing a tert-butoxycarbonyl group, the polarity of the molecule is reduced and solubility improved. Crucially, the steric and electronic effects of the protecting group significantly reduce the probability of unwanted bromination at the 2-methyl position, directing the reaction exclusively to the 6-position.
Q: Is this synthesis route suitable for large-scale manufacturing?
A: Yes, the process utilizes common industrial reagents like NBS and TFA, operates at moderate temperatures ranging from 0°C to 80°C, and employs standard organic solvents, making it highly amenable to commercial scale-up without requiring exotic catalysts or extreme conditions.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 6-(Dibromomethyl)-2-methylquinazoline-4(3H)-one Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-purity intermediates play in the development and manufacture of life-saving oncology therapies. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. We adhere to stringent purity specifications and utilize rigorous QC labs to guarantee that every batch of 6-(dibromomethyl)-2-methylquinazoline-4(3H)-one meets the exacting standards required for reference materials and API synthesis. Our commitment to quality ensures that your analytical methods remain robust and your final drug products are safe and effective.
We invite you to collaborate with us to optimize your supply chain for Raltitrexed and related quinazoline-based therapeutics. By leveraging our expertise in process chemistry, we can provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage potential partners to contact our technical procurement team to request specific COA data and route feasibility assessments. Together, we can ensure a reliable supply of this critical intermediate, supporting the global demand for high-quality cancer treatments while driving down manufacturing costs through superior process engineering.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →