Scalable Synthesis of Chiral Amino Alcohol Intermediates for Alzheimer's Therapeutics
Scalable Synthesis of Chiral Amino Alcohol Intermediates for Alzheimer's Therapeutics
The pharmaceutical industry constantly seeks robust methods for producing chirally pure intermediates, particularly for neurodegenerative disease treatments. Patent CN101472877A introduces a significant advancement in the preparation of 2S-amino alcohol intermediates, which are critical precursors for beta-amyloid protein inhibitors used in Alzheimer's disease therapy. This technology addresses the long-standing challenge of scaling asymmetric synthesis without compromising enantiomeric excess. By replacing hazardous azide reagents with a safer hydrazine-based reduction pathway, the process offers a viable route for reliable pharmaceutical intermediate supplier operations. The method ensures high stability and purity, essential for downstream drug substance manufacturing. This report analyzes the technical merits and commercial implications of this novel synthetic route for global supply chains.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional methods for synthesizing chiral amino acids and alcohols often rely on the use of trisyl azide or similar hazardous reagents to introduce nitrogen functionality. These conventional approaches frequently suffer from safety concerns associated with azide handling on a large scale, posing significant risks in commercial manufacturing environments. Furthermore, achieving high chiral purity often necessitates complex chromatographic separations, which drastically increase production costs and reduce overall throughput. The reliance on such reagents limits the ability to perform cost reduction in pharmaceutical intermediate manufacturing effectively. Additionally, the removal of byproducts from these reactions can be cumbersome, leading to lower yields and extended processing times. These factors collectively hinder the commercial scale-up of complex pharmaceutical intermediates required for modern therapeutics.
The Novel Approach
The patented methodology circumvents these issues by employing a di-tert-butyl diazene-1,2-dicarboxylate coupling strategy with an Evans oxazolidone chiral auxiliary. This approach eliminates the need for dangerous azide reagents, thereby enhancing operational safety and regulatory compliance within the facility. The process allows for the direct formation of the hydrazine intermediate which is subsequently reduced to the desired amino alcohol without the need for extensive purification columns. This streamlining of the synthetic route significantly simplifies the workflow and reduces the consumption of solvents and materials. Consequently, this novel approach facilitates reducing lead time for high-purity amino alcohols while maintaining stringent quality standards. The robustness of this chemistry makes it an attractive option for high-volume production.
Mechanistic Insights into Evans Oxazolidone Asymmetric Synthesis
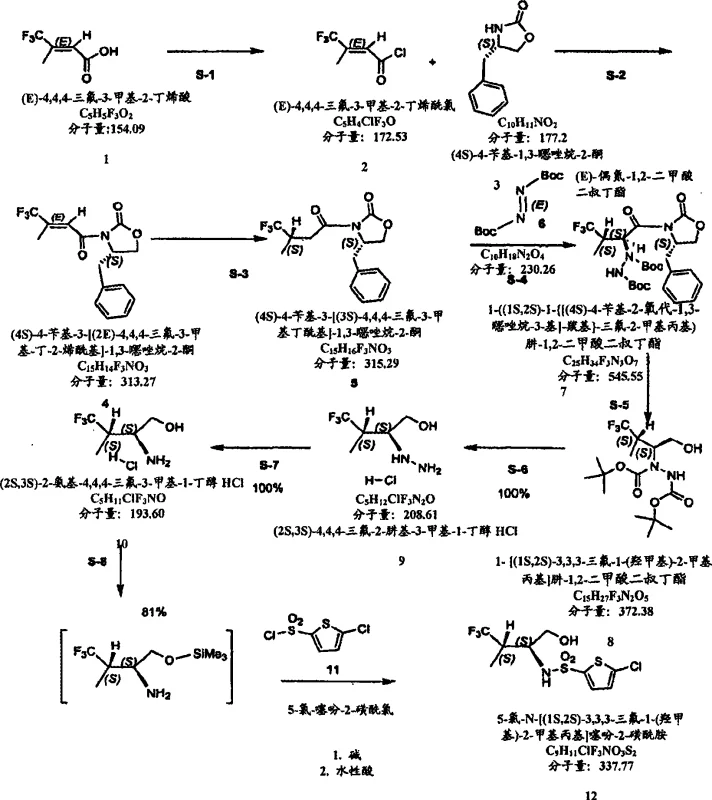
The core of this technology lies in the stereocontrolled acylation of the Evans oxazolidone auxiliary followed by an asymmetric azo-coupling reaction. The initial step involves the activation of a trifluoromethyl-substituted carboxylic acid, which is then coupled with (4S)-4-benzyl-1,3-oxazolidin-2-one to establish the first chiral center. Subsequent deprotonation with a strong base like LDA generates a Z-enolate that reacts stereoselectively with the azodicarboxylate. This step is critical as it sets the second chiral center with high diastereoselectivity, ensuring the formation of the desired 1S,2S configuration. The steric bulk of the oxazolidone ring directs the incoming electrophile to the less hindered face, minimizing the formation of unwanted diastereomers. This precise control is fundamental to achieving the high optical purity required for pharmaceutical applications.
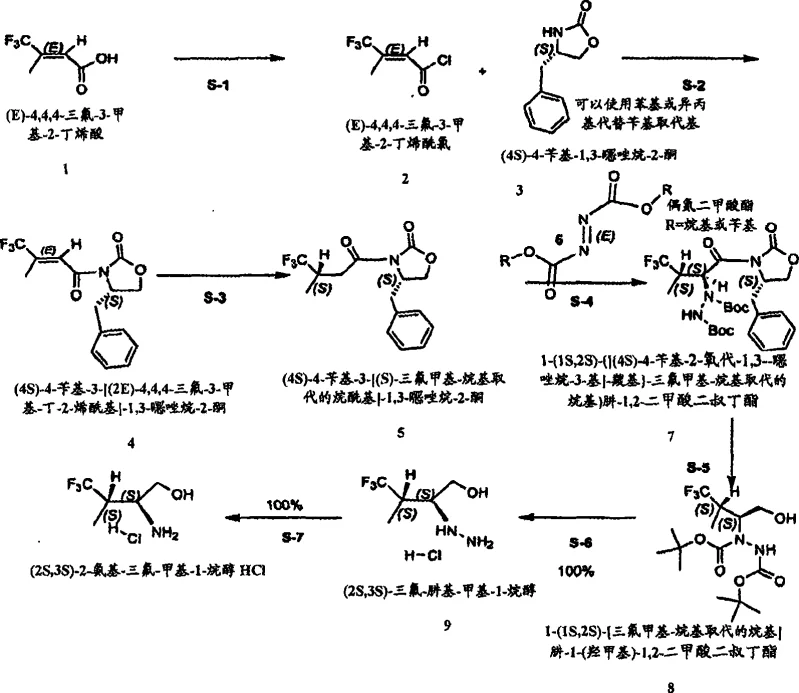
Following the coupling, the hydrazine dicarboxylate intermediate undergoes a reductive cleavage to release the chiral auxiliary and generate the hydrazino alcohol. This reduction is typically performed using lithium borohydride or similar reducing agents in a controlled solvent system. The resulting hydrazine is then subjected to acid-mediated deprotection to remove the Boc groups, yielding the hydrazine salt. Finally, catalytic hydrogenation over a metal catalyst such as PtO2 or Pd/C cleaves the N-N bond to furnish the target chiral amino alcohol hydrochloride. This sequence ensures that the chiral integrity established in the early steps is preserved throughout the synthesis. The mechanism avoids racemization pathways common in other methods, providing a consistent supply of high-purity material.
How to Synthesize Chiral Amino Alcohol Efficiently
Implementing this synthesis requires careful control of reaction temperatures and stoichiometry to maximize yield and purity. The process begins with the preparation of the acylated oxazolidone, followed by the low-temperature addition of the azodicarboxylate to maintain stereocontrol. Subsequent reduction and deprotection steps must be monitored to ensure complete conversion while avoiding side reactions. The final hydrogenation step is crucial for removing the hydrazine functionality and obtaining the free amine. Detailed standardized synthesis steps are provided below to guide process development teams in replicating this efficient route. Adhering to these parameters ensures the production of material suitable for further derivatization into active pharmaceutical ingredients.
- Activation of carboxylic acid and coupling with Evans oxazolidone to form the chiral acylated intermediate.
- Asymmetric azo-coupling using di-tert-butyl diazene-1,2-dicarboxylate to establish the second chiral center.
- Reductive cleavage and hydrogenation to yield the final chiral amino alcohol hydrochloride salt.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, this synthetic route offers substantial benefits regarding cost stability and supply security. By eliminating the need for expensive and hazardous azide reagents, the raw material costs are significantly optimized, contributing to overall cost reduction in pharmaceutical intermediate manufacturing. The process utilizes common solvents and catalysts that are readily available in the global chemical market, reducing the risk of supply chain disruptions. Furthermore, the avoidance of chromatographic purification steps lowers the consumption of silica gel and solvents, which are major cost drivers in fine chemical production. These efficiencies translate into a more competitive pricing structure for the final intermediate without compromising on quality. Supply chain managers can rely on this robust chemistry for consistent long-term sourcing.
- Cost Reduction in Manufacturing: The elimination of trisyl azide and chromatographic purification directly lowers the bill of materials and waste disposal costs. This streamlined process reduces the number of unit operations required, leading to lower labor and utility expenses per kilogram of product. The use of crystallization for purification instead of column chromatography significantly decreases solvent usage and processing time. These factors combine to create a highly cost-effective manufacturing process that enhances margin potential for downstream drug products. The economic advantages are derived from the inherent efficiency of the chemical transformations rather than arbitrary price cuts.
- Enhanced Supply Chain Reliability: The reagents used in this process, such as Evans oxazolidone and di-tert-butyl azodicarboxylate, are commercially available from multiple suppliers, ensuring supply continuity. The robustness of the reaction conditions means that production can be scaled up without significant re-optimization, reducing the risk of batch failures. This reliability is crucial for maintaining inventory levels and meeting the demanding schedules of pharmaceutical clients. By mitigating the risks associated with hazardous reagents, the process also reduces regulatory hurdles and inspection delays. This stability makes the supplier a more dependable partner for critical drug development programs.
- Scalability and Environmental Compliance: The process is designed with scale-up in mind, utilizing standard equipment like hydrogenation reactors and crystallizers that are common in chemical plants. The reduction in hazardous waste generation, particularly from azide byproducts, simplifies environmental compliance and waste treatment protocols. This aligns with modern green chemistry principles, making the manufacturing process more sustainable and environmentally friendly. The ability to handle large batches efficiently supports the transition from clinical trial material to commercial production volumes. This scalability ensures that the supply can grow in tandem with the market demand for the final therapeutic agent.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding this synthesis method. These answers are derived from the specific technical disclosures and advantages outlined in the patent documentation. Understanding these details helps stakeholders evaluate the feasibility of integrating this intermediate into their supply chain. The information provided clarifies the purity profiles, scalability, and regulatory aspects of the manufacturing process. This transparency supports informed decision-making for procurement and technical teams evaluating new suppliers.
Q: How does this method improve chiral purity compared to conventional routes?
A: The method utilizes Evans oxazolidone chiral auxiliaries which provide exceptional stereocontrol, avoiding the need for difficult chromatographic separations often required in traditional azide reduction methods.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the patent explicitly avoids reagents like trisyl azide that are unsuitable for scale-up, utilizing robust hydrogenation and crystallization steps that are amenable to multi-kilogram manufacturing.
Q: What are the key impurities controlled in this synthesis?
A: The process controls diastereomeric impurities through the chiral auxiliary and removes metal catalyst residues via specific solvent grinding and filtration steps, ensuring high chemical purity.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral Amino Alcohol Supplier
NINGBO INNO PHARMCHEM stands ready to leverage this advanced synthetic technology to support your drug development needs. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition smoothly from lab to plant. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch meets the highest industry standards. Our commitment to technical excellence allows us to deliver high-purity amino alcohol intermediates that facilitate your research and manufacturing goals. Partnering with us means accessing a supply chain that is both robust and responsive to your specific requirements.
We invite you to discuss how this technology can optimize your current sourcing strategy and reduce overall project costs. Our technical procurement team is available to provide a Customized Cost-Saving Analysis tailored to your specific volume needs. Please contact us to request specific COA data and route feasibility assessments for your target compounds. We are dedicated to supporting your success through reliable supply and technical expertise. Let us help you secure a stable supply of critical intermediates for your pharmaceutical pipeline.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →