Advanced Synthesis of Parecoxib Sodium: A Commercial Scale-Up Strategy for High-Purity Analgesic Intermediates
Advanced Synthesis of Parecoxib Sodium: A Commercial Scale-Up Strategy for High-Purity Analgesic Intermediates
The pharmaceutical industry continuously seeks robust manufacturing pathways for critical analgesic intermediates, particularly for COX-2 inhibitors like parecoxib sodium. Patent CN111100084A, published in May 2020, introduces a transformative preparation method that addresses longstanding challenges in the synthesis of this vital injectable pain management agent. Unlike traditional routes that rely on hazardous organolithium reagents or corrosive superacids, this novel approach utilizes 5-methyl-3,4-diphenyl isoxazole as a starting material and employs a sophisticated sequence of sulfonation, ammoniation, propionylation, and salification. The core innovation lies in the modulation of reaction kinetics using organic acids such as tartaric, citric, or formic acid, which creates a milder proton environment compared to conventional methods. This strategic adjustment not only enhances operational safety by drastically reducing the evolution of hydrogen chloride gas but also significantly improves the overall yield and purity profile of the final active pharmaceutical ingredient (API) precursor. For global procurement teams and R&D directors, this patent represents a pivotal shift towards more sustainable and economically viable manufacturing protocols for high-value pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historical synthesis routes for parecoxib sodium have been plagued by significant operational hazards and economic inefficiencies that hinder large-scale commercial adoption. As illustrated in prior art such as WO2003029230 and related literature, early methodologies frequently depended on the use of n-butyllithium for cyclization steps, a reagent known for its pyrophoric nature and extreme sensitivity to moisture, necessitating cryogenic conditions and specialized inert atmosphere equipment. Furthermore, dehydration steps often utilized trifluoroacetic acid (TFA), a superacid that imposes severe corrosion demands on reactor materials and generates fluorine-containing waste streams that are environmentally burdensome and costly to treat. Alternative routes disclosed in documents like WO2005123701 attempted to mitigate some issues by employing pyrrolidine condensation but introduced new complexities, such as the requirement for expensive acid-binding agents like 2,6-lutidine and prolonged reaction times extending up to 24 hours for chloroacetylation. These legacy processes result in fragmented supply chains, elevated capital expenditure for corrosion-resistant infrastructure, and inconsistent batch-to-batch purity due to the difficulty in controlling highly exothermic and sensitive reactions.
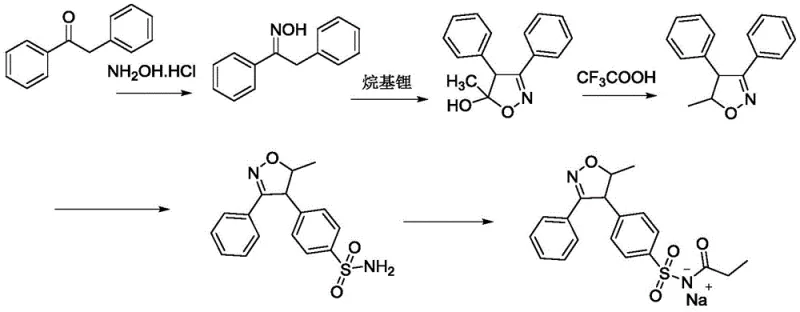
The Novel Approach
The methodology outlined in CN111100084A fundamentally reengineers the synthetic landscape by introducing a streamlined, three-stage process that prioritizes mild conditions and reagent accessibility. Instead of relying on aggressive superacids, the new protocol leverages the buffering capacity of organic acids—specifically tartaric, citric, or formic acid—to modulate the sulfonation reaction with chlorosulfonic acid. This subtle yet powerful modification allows the reaction to proceed efficiently at moderate temperatures between 10°C and 20°C, effectively suppressing side reactions and minimizing the generation of hazardous HCl gas. The subsequent propionylation step is equally innovative, utilizing a dichloromethane-acetonitrile mixed solvent system coupled with 4-pyrrolidinylpyridine as a nucleophilic catalyst. This catalytic system enables the acylation to occur rapidly at room temperature (20-25°C), eliminating the need for thermal activation or extended reaction durations. By simplifying the workflow to essentially sulfonation, ammoniation, and propionylation followed by direct salification, the process removes multiple purification bottlenecks and protective group manipulations found in older patents, thereby offering a direct path to cost reduction in pharmaceutical intermediates manufacturing while ensuring a robust impurity profile suitable for parenteral applications.
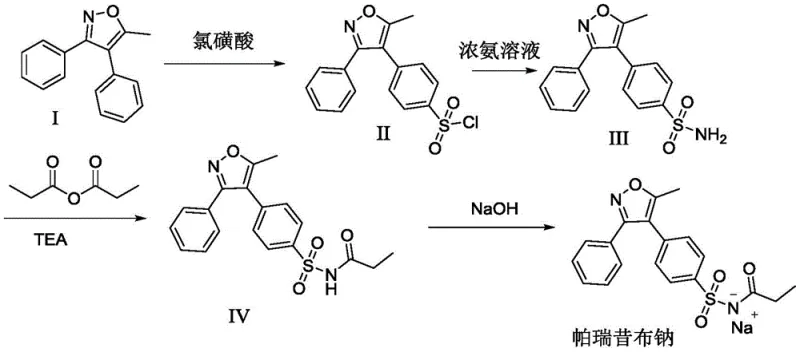
Mechanistic Insights into Organic Acid Modulated Sulfonation
The mechanistic elegance of this patented process centers on the precise control of electrophilic aromatic substitution during the sulfonation of the isoxazole core. In traditional superacid media, the high proton activity can lead to over-sulfonation or degradation of the sensitive isoxazole ring. By introducing organic acids like tartaric acid at a molar ratio of 1.5-2:1 relative to the substrate, the reaction medium achieves a buffered acidity that facilitates the formation of the sulfonyl chloride intermediate without compromising the structural integrity of the heterocycle. The addition of sodium iodide acts as a crucial promoter, likely functioning through a halogen exchange mechanism or by enhancing the nucleophilicity of the aromatic system, which drives the equilibrium towards the desired product. This synergistic effect accelerates the reaction rate, allowing completion within approximately 1 hour at 40°C, a significant improvement over the multi-hour timelines of conventional methods. Furthermore, the use of alkali bases such as sodium carbonate or potassium carbonate in the subsequent neutralization step ensures that the acidic byproducts are sequestered effectively, preventing acid-catalyzed hydrolysis of the newly formed sulfonamide bond during the ammoniation phase.
Impurity control is inherently built into this mechanistic framework through the selection of reagents and solvent systems. The avoidance of strong Lewis acids and pyrophoric reagents eliminates the formation of metal-complexed impurities that are notoriously difficult to remove to ppm levels required for injectables. The propionylation step, catalyzed by 4-pyrrolidinylpyridine, exhibits high chemoselectivity for the sulfonamide nitrogen, minimizing O-acylation or ring-opening side reactions that could generate genotoxic impurities. The final crystallization from methanol serves as a potent purification gate, leveraging the differential solubility of the sodium salt to exclude unreacted starting materials and organic byproducts. Analytical data from the patent examples demonstrates that this approach consistently yields parecoxib sodium with purity exceeding 99.8% and single impurity levels below 0.1%, meeting the stringent specifications demanded by regulatory bodies for high-purity pharmaceutical intermediates.
How to Synthesize Parecoxib Sodium Efficiently
The synthesis of parecoxib sodium via this patented route offers a practical blueprint for laboratories and pilot plants aiming to establish a reliable supply chain for this critical analgesic. The process is characterized by its operational simplicity, utilizing standard glass-lined or stainless steel reactors without the need for exotic metallurgy. The initial sulfonation requires careful temperature control during the dropwise addition of chlorosulfonic acid to manage exotherms, followed by a straightforward aqueous workup and extraction. The subsequent propionylation is remarkably forgiving, proceeding to completion in just 2 hours at ambient temperature, which simplifies energy management and scheduling. Detailed standardized operating procedures for scaling this chemistry from gram to kilogram quantities are essential for maintaining the high purity profiles observed in the patent examples. For a comprehensive breakdown of the specific stoichiometric ratios, temperature ramps, and workup protocols, please refer to the technical guide below.
- Sulfonation and Ammoniation: React 5-methyl-3,4-diphenyl isoxazole with chlorosulfonic acid in the presence of tartaric, citric, or formic acid at 10-20°C, followed by neutralization and reaction with concentrated ammonia to form the sulfonamide intermediate.
- Propionylation: Dissolve the sulfonamide intermediate in a dichloromethane-acetonitrile mixed solvent, add 4-pyrrolidinylpyridine as a catalyst, and react with propionic anhydride and triethylamine at 20-25°C.
- Salification: Dissolve the propionylated product in methanol, add sodium hydroxide at 50°C, and crystallize the final parecoxib sodium salt by cooling to 0-5°C.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthesis route translates directly into enhanced operational resilience and significant cost optimization opportunities. The elimination of n-butyllithium and trifluoroacetic acid removes two of the most volatile cost drivers in fine chemical manufacturing, both in terms of raw material expense and the specialized handling infrastructure they require. By shifting to commodity organic acids and stable amine catalysts, the process insulates the supply chain from the price fluctuations associated with specialty organometallic reagents. Furthermore, the mild reaction conditions reduce the wear and tear on production equipment, extending asset life and lowering maintenance downtime. The simplified workflow, which avoids complex protection-deprotection sequences, shortens the overall cycle time per batch, thereby increasing throughput capacity without the need for additional capital investment in reactor volume. These factors collectively contribute to a more predictable and economical manufacturing model for high-volume API production.
- Cost Reduction in Manufacturing: The replacement of expensive and hazardous reagents like n-butyllithium and 2,6-lutidine with cost-effective organic acids and sodium salts dramatically lowers the bill of materials. Additionally, the reduction in reaction time and the ability to operate at ambient temperatures significantly decrease energy consumption for heating and cooling, leading to substantial operational expenditure savings. The high yield reported in the patent examples minimizes raw material waste, further optimizing the cost per kilogram of the final active ingredient.
- Enhanced Supply Chain Reliability: Sourcing stability is improved by relying on widely available bulk chemicals rather than niche reagents that may face supply constraints. The robustness of the process against minor variations in temperature and mixing ensures consistent batch quality, reducing the risk of production delays caused by out-of-specification results. This reliability is critical for maintaining continuous supply to downstream formulation partners, especially for injectable products where inventory buffers are often kept lean.
- Scalability and Environmental Compliance: The process is inherently scalable, having been designed with industrial production in mind. The reduction in hazardous waste generation, particularly the avoidance of fluorine-containing effluents and lithium salts, simplifies wastewater treatment and lowers environmental compliance costs. The use of common solvents like dichloromethane and acetonitrile facilitates efficient solvent recovery and recycling, aligning with modern green chemistry principles and corporate sustainability goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis method. Understanding these nuances is vital for technology transfer teams and quality assurance personnel evaluating the feasibility of adopting this route for commercial manufacture. The answers are derived directly from the experimental data and technical disclosures within the patent specification, providing a factual basis for decision-making.
Q: How does the new method improve upon traditional n-butyllithium routes?
A: The new method replaces hazardous and expensive n-butyllithium and superacid trifluoroacetic acid with mild organic acids (tartaric, citric) and sodium iodide. This significantly reduces equipment corrosion risks, eliminates the need for cryogenic conditions, and lowers raw material costs while maintaining high purity (>99%).
Q: What are the critical quality parameters for the sulfonation step?
A: Critical parameters include maintaining the reaction temperature between 10-20°C during chlorosulfonic acid addition to prevent over-sulfonation or degradation. The molar ratio of organic acid to substrate (1.5-2:1) and the subsequent addition of sodium iodide are crucial for driving the equilibrium forward and minimizing HCl gas generation.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process is specifically designed for industrial scalability. It operates at mild temperatures (20-50°C), uses common solvents like dichloromethane and acetonitrile, and avoids pyrophoric reagents. The simplified workup procedures, such as direct crystallization from methanol, facilitate efficient commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Parecoxib Sodium Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of securing a stable and high-quality supply of parecoxib sodium for the global pharmaceutical market. Our technical team has thoroughly analyzed the innovations presented in CN111100084A and possesses the expertise to implement this advanced organic acid-catalyzed route at an industrial scale. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to full-scale manufacturing is seamless and compliant with cGMP standards. Our state-of-the-art facilities are equipped with corrosion-resistant reactors and rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch meets the exacting requirements for parenteral drug substances.
We invite potential partners to engage with our technical procurement team to discuss how this optimized synthesis route can benefit your specific supply chain needs. By leveraging our process development capabilities, we can provide a Customized Cost-Saving Analysis tailored to your volume requirements, demonstrating the tangible economic advantages of switching to this greener, more efficient methodology. We encourage you to contact us to request specific COA data from our pilot batches and detailed route feasibility assessments, allowing you to make informed decisions that enhance both the quality and profitability of your analgesic product portfolio.