Advanced Copper-Catalyzed Synthesis of Pyridine Derivatives for Commercial Scale-Up
Advanced Copper-Catalyzed Synthesis of Pyridine Derivatives for Commercial Scale-Up
The pharmaceutical industry continuously seeks robust and cost-effective synthetic routes for critical kinase inhibitor intermediates, particularly those targeting Cell Cycle Dependent Kinases (CDKs). Patent CN115521289A introduces a transformative methodology for synthesizing pyridine derivatives, specifically focusing on the production of tert-butyl 4-(6-aminopyridin-3-yl)piperidine-1-carboxylate, a pivotal building block for CDK4/6 inhibitors. This disclosure addresses the longstanding economic and technical bottlenecks associated with traditional precious metal catalysis by leveraging a copper-mediated Grignard coupling strategy. For R&D directors and procurement specialists, this shift represents a significant opportunity to optimize supply chains for high-purity pharmaceutical intermediates. The technology not only enhances the purity profile of the final product but also drastically simplifies the downstream processing required to meet stringent regulatory standards for residual metals in active pharmaceutical ingredients.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of key pyridine-piperidine scaffolds has relied heavily on palladium-catalyzed cross-coupling reactions, which, while effective, impose substantial burdens on manufacturing economics and environmental compliance. As illustrated in the prior art pathways, methods such as those disclosed in CN109384767A utilize catalysts like Pd2(dba)3 alongside complex ligand systems like Xphos to couple N-benzyl piperidone with brominated pyridine derivatives. 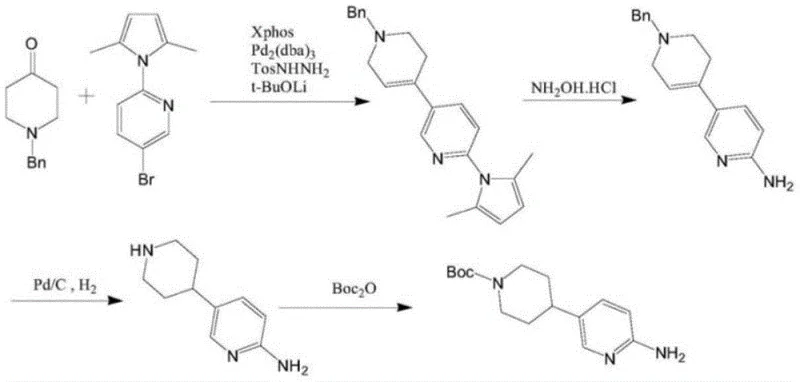 These conventional approaches suffer from the high intrinsic cost of palladium complexes, which are subject to volatile market pricing and supply constraints. Furthermore, the removal of residual palladium from the final API intermediate is a technically demanding and costly purification step, often requiring specialized scavengers or multiple recrystallization cycles that erode overall yield. The reliance on benzyl protecting groups also necessitates additional hydrogenation steps, adding complexity and safety risks associated with high-pressure hydrogen handling in early synthetic stages.
These conventional approaches suffer from the high intrinsic cost of palladium complexes, which are subject to volatile market pricing and supply constraints. Furthermore, the removal of residual palladium from the final API intermediate is a technically demanding and costly purification step, often requiring specialized scavengers or multiple recrystallization cycles that erode overall yield. The reliance on benzyl protecting groups also necessitates additional hydrogenation steps, adding complexity and safety risks associated with high-pressure hydrogen handling in early synthetic stages.
The Novel Approach
In stark contrast, the methodology presented in CN115521289A circumvents these issues by employing a copper-catalyzed coupling of a halogenated pyridine derivative with a Grignard reagent in the presence of di-tert-butyl dicarbonate. 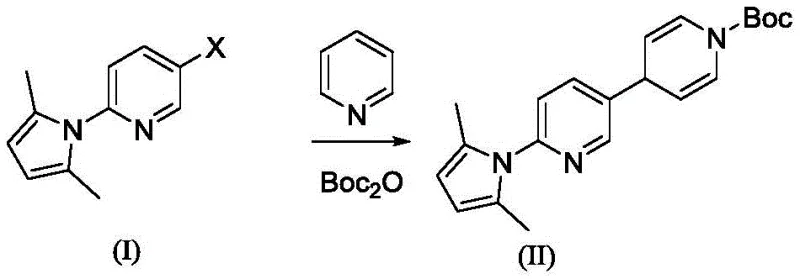 This innovative route utilizes inexpensive copper salts, such as CuI or CuBr, which are orders of magnitude cheaper than palladium alternatives and do not require sophisticated ligand systems. The process operates under mild temperatures ranging from -20°C to 50°C, typically optimized between 0°C and 30°C, ensuring excellent thermal safety profiles suitable for large-scale reactors. By directly installing the Boc protecting group during the coupling phase, the synthesis eliminates the need for separate protection and deprotection sequences found in older routes. This streamlined approach not only reduces the total number of unit operations but also minimizes solvent consumption and waste generation, aligning perfectly with green chemistry principles and cost reduction in pharmaceutical intermediate manufacturing.
This innovative route utilizes inexpensive copper salts, such as CuI or CuBr, which are orders of magnitude cheaper than palladium alternatives and do not require sophisticated ligand systems. The process operates under mild temperatures ranging from -20°C to 50°C, typically optimized between 0°C and 30°C, ensuring excellent thermal safety profiles suitable for large-scale reactors. By directly installing the Boc protecting group during the coupling phase, the synthesis eliminates the need for separate protection and deprotection sequences found in older routes. This streamlined approach not only reduces the total number of unit operations but also minimizes solvent consumption and waste generation, aligning perfectly with green chemistry principles and cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Copper-Catalyzed Grignard Coupling
The core of this technological advancement lies in the precise orchestration of organometallic chemistry, where a Grignard reagent, typically isopropylmagnesium chloride or cyclohexylmagnesium chloride, acts as the nucleophilic driver. The mechanism initiates with the formation of the Grignard species under strictly anhydrous conditions, utilizing solvents like tetrahydrofuran (THF) or diethyl ether to stabilize the organomagnesium complex. Upon introduction of the copper catalyst, a transmetallation event occurs, generating a reactive organocopper species that is significantly more selective than the corresponding Grignard reagent alone. This organocopper intermediate then attacks the electrophilic center of the halogenated pyridine substrate, facilitating the carbon-carbon bond formation with high regioselectivity. The presence of di-tert-butyl dicarbonate (Boc2O) in the reaction mixture serves a dual purpose: it traps the nitrogen atom of the pyridine ring or the incoming nucleophile to form the stable carbamate protection immediately, preventing over-reaction or polymerization side products that often plague uncatalyzed Grignard additions.
From an impurity control perspective, this mechanism offers superior manageability compared to palladium cycles. The absence of palladium eliminates the risk of forming difficult-to-remove metal-organic complexes that can persist through multiple purification stages. Instead, the copper residues are generally more water-soluble or can be easily chelated and removed during the aqueous workup phase, which involves quenching with ammonium chloride solution followed by extraction. The reaction conditions are tuned to minimize the formation of homocoupling byproducts, a common issue in Grignard chemistry, by maintaining a controlled stoichiometry where the copper catalyst loading is kept between 0.01 to 1 times the mass of the substrate. This precise control ensures that the resulting intermediate, such as the compound of formula (II), achieves high HPLC purity levels exceeding 97% directly after crystallization, reducing the burden on downstream purification teams and ensuring a consistent quality profile for subsequent synthetic steps.
How to Synthesize tert-butyl 4-(6-aminopyridin-3-yl)piperidine-1-carboxylate Efficiently
The practical execution of this synthesis involves a sequential workflow designed for maximum operational efficiency and safety in a GMP environment. The process begins with the preparation of the Grignard reagent and its subsequent coupling with the pyridine precursor to establish the core carbon skeleton with the Boc group intact. Following this, a catalytic hydrogenation step reduces the aromatic pyridine ring to the saturated piperidine system, a transformation that is critical for the biological activity of the final CDK inhibitor. The final stage involves the selective removal of the 2,5-dimethylpyrrolyl protecting group using hydroxylamine hydrochloride under basic conditions, yielding the free amine functionality required for the final coupling with the kinase inhibitor core. This modular approach allows for easy isolation of intermediates, enabling rigorous quality control checks at each stage to ensure the integrity of the supply chain.
- Perform a copper-catalyzed coupling of a halogenated pyridine derivative with a Grignard reagent and di-tert-butyl dicarbonate to form the protected intermediate.
- Execute catalytic hydrogenation using Pd/C or Pd(OH)2/C in an alkaline environment to reduce the pyridine ring to a piperidine structure.
- Remove the 2,5-dimethylpyrrolyl protecting group using hydroxylamine hydrochloride under basic conditions to yield the final amine product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this copper-catalyzed route offers compelling strategic advantages that extend beyond simple raw material savings. The shift away from precious metals fundamentally alters the cost structure of the manufacturing process, removing exposure to the volatile pricing of palladium and rhodium markets. By utilizing base metals like copper, which are abundant and geographically diverse in supply, manufacturers can secure long-term supply contracts with greater stability and predictability. This resilience is crucial for maintaining continuous production schedules for high-demand oncology therapeutics, where supply interruptions can have severe clinical and financial consequences. Furthermore, the simplified workup procedures reduce the consumption of specialized scavenging resins and chromatography media, leading to substantial cost savings in consumables and waste disposal fees.
- Cost Reduction in Manufacturing: The elimination of expensive palladium catalysts and complex phosphine ligands results in a drastic reduction in direct material costs per kilogram of product. Additionally, the streamlined process reduces the number of isolation and purification steps, which lowers labor costs and energy consumption associated with solvent evaporation and drying. The ability to achieve high purity without extensive chromatographic purification further enhances the economic viability of the process, making it highly competitive for generic and branded drug production alike.
- Enhanced Supply Chain Reliability: The reagents required for this synthesis, such as CuI, Boc2O, and standard Grignard reagents, are commodity chemicals available from multiple global suppliers, reducing the risk of single-source dependency. The robustness of the reaction conditions, which tolerate a wider range of temperatures and do not require ultra-low temperature cryogenic cooling, simplifies facility requirements and allows for production in a broader range of manufacturing sites. This flexibility ensures that production can be scaled up or shifted between facilities without significant re-validation efforts, securing the supply continuity for critical pharmaceutical intermediates.
- Scalability and Environmental Compliance: The process generates significantly less hazardous waste compared to traditional methods, as it avoids the use of toxic heavy metals that require specialized disposal protocols. The aqueous workup and crystallization steps are inherently scalable, allowing for seamless transition from pilot plant batches to multi-ton commercial production. This alignment with environmental, social, and governance (ESG) goals not only reduces regulatory compliance burdens but also enhances the sustainability profile of the final drug product, a factor increasingly valued by healthcare providers and patients.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthetic route. These insights are derived directly from the experimental data and process descriptions within the patent documentation, providing a clear understanding of the operational realities. Understanding these nuances is essential for technical teams evaluating the feasibility of integrating this chemistry into their existing manufacturing portfolios.
Q: How does the copper-catalyzed route improve cost efficiency compared to palladium methods?
A: The novel process replaces expensive palladium catalysts like Pd2(dba)3 with economical copper iodide (CuI), significantly reducing raw material costs and eliminating complex heavy metal removal steps required for API compliance.
Q: What are the critical reaction conditions for the Grignard coupling step?
A: The reaction requires strict anhydrous conditions using solvents like THF under inert gas protection, with temperatures maintained between 0°C and 30°C to ensure optimal yield and minimize side reactions.
Q: Is this synthesis route suitable for large-scale commercial production?
A: Yes, the process utilizes robust reagents and avoids sensitive precious metal catalysts, making it highly scalable for industrial manufacturing while maintaining high purity standards for pharmaceutical applications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable tert-butyl 4-(6-aminopyridin-3-yl)piperidine-1-carboxylate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthetic routes in the development of next-generation oncology therapies. Our team of expert chemists has extensively evaluated the copper-catalyzed methodology described in CN115521289A and possesses the technical capability to implement this process at an industrial scale. We offer extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with consistency and reliability. Our state-of-the-art facilities are equipped with rigorous QC labs capable of detecting trace impurities and ensuring stringent purity specifications that exceed international pharmacopeial standards. We are committed to delivering high-quality intermediates that facilitate the rapid advancement of your drug development programs.
We invite you to collaborate with us to optimize your supply chain for CDK4/6 inhibitor intermediates. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. By leveraging our expertise in process optimization and scale-up, we can help you reduce lead times and lower overall manufacturing costs. Please contact us to request specific COA data and route feasibility assessments, and let us demonstrate how our advanced manufacturing capabilities can support your strategic goals in the pharmaceutical market.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →