Advanced Synthesis of Tricyclic Alkylhydroxamates for Next-Generation Oncology Therapeutics
Advanced Synthesis of Tricyclic Alkylhydroxamates for Next-Generation Oncology Therapeutics
The pharmaceutical landscape for oncology treatments is continuously evolving, driven by the need for more potent and selective epigenetic modulators. Patent CN1507441A introduces a groundbreaking class of tricyclic alkylhydroxamates that exhibit exceptional efficacy as histone deacetylase (HDAC) inhibitors. Unlike traditional hydroxamic acid derivatives, these novel compounds incorporate complex tricyclic scaffolds such as dibenzo[b,e]oxepin, fluorene, and carbazole systems, which significantly enhance their binding affinity and biological activity. The patent details comprehensive synthetic methodologies that allow for the precise construction of these molecules, ensuring high purity and structural integrity essential for clinical applications. 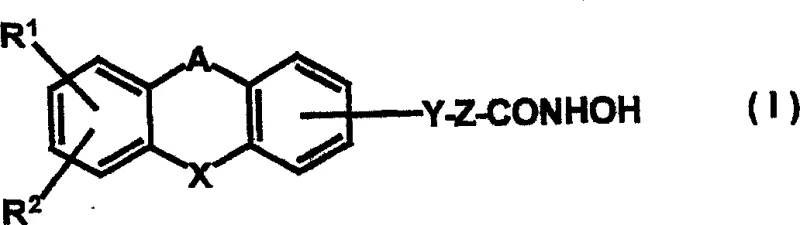 This technological advancement represents a critical leap forward for reliable pharmaceutical intermediate supplier networks aiming to support the development of next-generation antitumor agents. The disclosed compounds have demonstrated superior cell proliferation inhibition compared to existing standards, positioning them as high-value candidates for drug discovery pipelines focused on colorectal carcinoma, T-cell lymphoma, and other malignancies.
This technological advancement represents a critical leap forward for reliable pharmaceutical intermediate supplier networks aiming to support the development of next-generation antitumor agents. The disclosed compounds have demonstrated superior cell proliferation inhibition compared to existing standards, positioning them as high-value candidates for drug discovery pipelines focused on colorectal carcinoma, T-cell lymphoma, and other malignancies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of hydroxamic acid-based HDAC inhibitors has faced significant challenges regarding stability, selectivity, and overall yield. Conventional routes often rely on simple aromatic or aliphatic linkers that fail to provide the necessary steric bulk and electronic properties required for optimal enzyme interaction. Furthermore, traditional methods frequently struggle with the direct introduction of the hydroxamic acid moiety due to its sensitivity to hydrolysis and oxidation, often necessitating complex multi-step protection strategies that degrade process efficiency. Many prior art processes utilize harsh reaction conditions that can compromise the integrity of sensitive tricyclic cores, leading to impurity profiles that are difficult to manage during scale-up. The lack of robust methodologies for coupling bulky tricyclic phenols with long-chain hydroxamic acid precursors has limited the exploration of this chemical space, resulting in a scarcity of diverse, high-potency candidates for clinical evaluation.
The Novel Approach
The methodology outlined in CN1507441A overcomes these historical barriers through a sophisticated yet practical synthetic strategy centered on orthogonal protection and mild coupling conditions. By employing a benzyl-protected hydroxylamine intermediate, the process safeguards the sensitive hydroxamic acid functionality during the critical etherification step with the tricyclic core. This approach allows for the use of elevated temperatures and strong bases to drive the coupling reaction to completion without degrading the final pharmacophore. 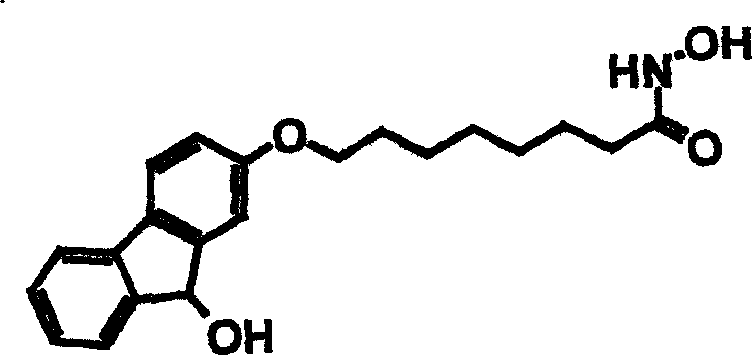 The versatility of this novel approach is evident in its ability to accommodate diverse tricyclic systems, including fluorene and carbazole derivatives, as shown in the successful synthesis of racemic-8-(9-hydroxy-9H-fluoren-2-yloxy)-suberoyl hydroxamic acid. This flexibility enables rapid structure-activity relationship (SAR) studies, facilitating the identification of lead compounds with optimized pharmacokinetic properties. The final deprotection step utilizes catalytic hydrogenolysis, a clean and scalable technique that avoids the generation of hazardous waste streams associated with acidic or basic hydrolysis, thereby aligning with modern green chemistry principles in cost reduction in pharmaceutical intermediates manufacturing.
The versatility of this novel approach is evident in its ability to accommodate diverse tricyclic systems, including fluorene and carbazole derivatives, as shown in the successful synthesis of racemic-8-(9-hydroxy-9H-fluoren-2-yloxy)-suberoyl hydroxamic acid. This flexibility enables rapid structure-activity relationship (SAR) studies, facilitating the identification of lead compounds with optimized pharmacokinetic properties. The final deprotection step utilizes catalytic hydrogenolysis, a clean and scalable technique that avoids the generation of hazardous waste streams associated with acidic or basic hydrolysis, thereby aligning with modern green chemistry principles in cost reduction in pharmaceutical intermediates manufacturing.
Mechanistic Insights into Benzyl-Protection and Etherification Strategy
The core of this synthetic innovation lies in the strategic use of O-benzylhydroxylamine as a masked hydroxamic acid equivalent. In the initial activation step, omega-bromo alkanoic acids are coupled with O-benzylhydroxylamine using phosphinic chloride activators, forming a stable amide bond that withstands subsequent nucleophilic attacks. This protection group is chemically robust yet easily removable under mild hydrogenolytic conditions using palladium catalysts, ensuring that the final product is obtained in high purity without residual metal contaminants. The subsequent etherification involves a nucleophilic aromatic substitution or SN2-type displacement where the phenolic oxygen of the tricyclic core attacks the alkyl bromide of the protected chain. 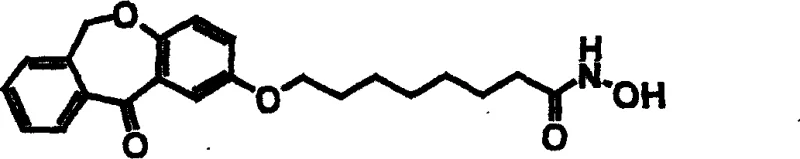 This step is critical for establishing the correct spatial orientation of the zinc-binding group relative to the cap group, which is essential for fitting into the narrow tunnel of the HDAC enzyme active site. The patent demonstrates that using polar aprotic solvents like DMF combined with inorganic bases like potassium carbonate facilitates this transformation efficiently, even with sterically hindered substrates. The mechanistic precision ensures that the resulting tricyclic alkylhydroxamates maintain the correct stereochemistry and chain length required for potent biological activity, minimizing the formation of regioisomers that could complicate downstream purification.
This step is critical for establishing the correct spatial orientation of the zinc-binding group relative to the cap group, which is essential for fitting into the narrow tunnel of the HDAC enzyme active site. The patent demonstrates that using polar aprotic solvents like DMF combined with inorganic bases like potassium carbonate facilitates this transformation efficiently, even with sterically hindered substrates. The mechanistic precision ensures that the resulting tricyclic alkylhydroxamates maintain the correct stereochemistry and chain length required for potent biological activity, minimizing the formation of regioisomers that could complicate downstream purification.
Impurity control is inherently built into this process design through the crystallinity and stability of the protected intermediates. The benzyl-protected precursors often exhibit favorable physical properties that allow for purification via column chromatography or crystallization before the final deprotection step. This 'purify-before-deprotect' strategy is vital for high-purity pharmaceutical intermediates production, as it prevents the co-elution of unreacted starting materials or side products with the final polar hydroxamic acid. Additionally, the use of specific catalysts like palladium on barium carbonate or barium sulfate for the hydrogenolysis step minimizes over-reduction of the tricyclic core, preserving the integrity of ketone or double bond functionalities within the scaffold. Rigorous quality control at the intermediate stage ensures that the final API precursor meets stringent specifications for heavy metals and organic impurities, a key requirement for regulatory approval in oncology drug development.
How to Synthesize Tricyclic Alkylhydroxamates Efficiently
The synthesis of these high-value compounds follows a logical progression designed for reproducibility and scalability in a GMP environment. The process begins with the preparation of the protected hydroxamic acid chain, followed by coupling with the tricyclic phenol, and concludes with global deprotection. Detailed operational parameters, including solvent choices, temperature ranges, and stoichiometric ratios, are optimized to maximize yield while minimizing waste. For a comprehensive breakdown of the standardized operating procedures and safety protocols required for this synthesis, please refer to the technical guide below.
- Activation and Protection: React O-benzylhydroxylamine hydrochloride with omega-bromo suberic acid using bis(2-oxo-3-oxazolidinyl)phosphinic chloride to form the protected benzyl hydroxamate intermediate.
- Etherification Coupling: Couple the protected hydroxamate with 2-hydroxy-6H-dibenzo[b,e]oxepin-11-one in DMF using potassium carbonate at elevated temperatures (100°C) to form the tricyclic ether linkage.
- Catalytic Deprotection: Perform hydrogenolysis using palladium on barium carbonate in methanol to remove the benzyl protecting group, yielding the final free hydroxamic acid.
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain perspective, the synthetic route described in CN1507441A offers substantial advantages in terms of raw material availability and process robustness. The starting materials, such as bromo-alkanoic acids and various tricyclic ketones or phenols, are commodity chemicals available from multiple global suppliers, reducing the risk of single-source bottlenecks. The reliance on standard unit operations like filtration, extraction, and distillation means that the process can be easily transferred to existing multipurpose manufacturing facilities without requiring specialized equipment investments. This adaptability significantly enhances supply chain reliability, ensuring consistent delivery schedules for downstream drug manufacturers who depend on timely access to critical intermediates for their clinical trials and commercial launches.
- Cost Reduction in Manufacturing: The elimination of exotic reagents and the use of catalytic hydrogenolysis for deprotection contribute to a more economical process profile. By avoiding stoichiometric amounts of expensive coupling reagents in the final step and utilizing recoverable palladium catalysts, the overall cost of goods sold (COGS) is optimized. Furthermore, the high purity of intermediates reduces the burden on downstream purification, lowering solvent consumption and waste disposal costs. This efficiency translates into significant cost savings for partners seeking a reliable pharmaceutical intermediate supplier capable of delivering competitive pricing without compromising quality.
- Enhanced Supply Chain Reliability: The modular nature of the synthesis allows for the decoupling of production stages. The protected hydroxamic acid chains can be manufactured in bulk and stored stably, ready for coupling with different tricyclic cores as demand dictates. This flexibility mitigates the risk of production delays caused by the shortage of a specific tricyclic building block. Additionally, the robustness of the chemical transformations ensures high batch-to-batch consistency, reducing the likelihood of failed batches that could disrupt the supply of commercial scale-up of complex pharmaceutical intermediates.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing solvents and conditions that are amenable to large-scale reactor operations. The final hydrogenolysis step generates benign byproducts (toluene and water), simplifying effluent treatment and ensuring compliance with increasingly strict environmental regulations. The ability to scale from kilogram to multi-ton quantities while maintaining product quality makes this technology ideal for meeting the growing global demand for epigenetic therapies. This scalability supports reducing lead time for high-purity pharmaceutical intermediates, enabling faster time-to-market for new oncology drugs.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these tricyclic hydroxamates. The answers are derived directly from the experimental data and claims presented in the patent documentation, providing clarity on the feasibility and performance of this technology. Understanding these details is crucial for stakeholders evaluating the potential integration of these compounds into their development portfolios.
Q: What is the primary biological mechanism of these tricyclic compounds?
A: These compounds function as histone deacetylase (HDAC) inhibitors. By blocking HDAC enzymes, they increase histone acetylation, leading to relaxed chromatin structure and reactivation of tumor suppressor genes, thereby inhibiting cell proliferation and inducing apoptosis in cancer cells.
Q: How does the potency of these novel hydroxamates compare to standard references like SAHA?
A: According to the patent data, specific embodiments demonstrate significantly higher inhibitory activity. For instance, Example 5 showed 100% inhibition at 10nM concentration, whereas the reference compound Suberanilohydroxamic Acid (SAHA) showed only 42% inhibition under identical testing conditions.
Q: Are the starting materials for this synthesis commercially viable for scale-up?
A: Yes, the synthesis utilizes readily available commodity chemicals such as bromo-alkanoic acids, substituted phenols (like carbazole or fluorenone derivatives), and standard protecting group reagents. The process avoids exotic catalysts, relying on standard base-mediated coupling and palladium-catalyzed hydrogenolysis.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tricyclic Alkylhydroxamates Supplier
NINGBO INNO PHARMCHEM stands at the forefront of custom synthesis, leveraging deep expertise in complex organic transformations to bring patented technologies like CN1507441A to life. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and reliability. We operate state-of-the-art rigorous QC labs that enforce stringent purity specifications, guaranteeing that every batch of tricyclic alkylhydroxamates meets the highest industry standards for epigenetic research and drug development. Our commitment to quality assurance ensures that the intricate structural requirements of these HDAC inhibitors are maintained throughout the manufacturing process.
We invite you to collaborate with us to unlock the full potential of this innovative chemistry. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can accelerate your project timelines. Let us be your partner in advancing the next generation of cancer therapeutics through superior chemical execution and supply chain excellence.