Scalable Synthesis of Imidazo[1,2-a][1,8]-naphthyridine Amides for Next-Gen Antiviral Therapies
Scalable Synthesis of Imidazo[1,2-a][1,8]-naphthyridine Amides for Next-Gen Antiviral Therapies
The global pharmaceutical landscape is continuously evolving to address persistent viral challenges, with Hepatitis C Virus (HCV) remaining a critical focus area despite recent advancements. Patent CN103333168A introduces a significant breakthrough in the chemical space of antiviral agents by disclosing a novel class of amide compounds based on the imidazo[1,2-a][1,8]-naphthyridine scaffold. This intellectual property outlines a robust and efficient preparation method that transforms simple, commercially accessible raw materials into highly active HCV inhibitors through a concise four-to-five-step sequence. For R&D directors and procurement specialists, this represents a pivotal opportunity to secure reliable pharmaceutical intermediate supplier partnerships that can deliver high-purity scaffolds essential for developing next-generation, interferon-free therapeutic regimens. The technical depth of this patent suggests a pathway that balances molecular complexity with manufacturing feasibility, addressing the urgent need for drugs with higher therapeutic indices and reduced side effect profiles compared to traditional protease inhibitors.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of complex heterocyclic systems like naphthyridines often involved multi-step sequences requiring harsh reaction conditions, toxic reagents, or expensive transition metal catalysts that complicate downstream processing. Conventional routes frequently suffered from low atom economy and generated significant amounts of hazardous waste, posing challenges for environmental compliance and cost reduction in API manufacturing. Furthermore, many traditional methods relied on specialized starting materials that were not readily available on a commercial scale, leading to supply chain bottlenecks and extended lead times for high-purity pharmaceutical intermediates. The reliance on heavy metal catalysts also necessitated rigorous purification steps to meet stringent residual metal specifications, adding both time and expense to the overall production cycle. These factors collectively hindered the rapid scale-up of promising antiviral candidates, delaying their entry into clinical trials and eventual market availability for patients in need of effective HCV treatments.
The Novel Approach
In stark contrast, the methodology described in the patent leverages a streamlined synthetic strategy that begins with ubiquitous building blocks such as 2,6-diaminopyridine and substituted diketones. This novel approach eliminates the need for precious metal catalysis in the core ring-forming steps, utilizing phosphoric acid as a benign and effective promoter for the initial cyclization. The subsequent construction of the imidazole ring via condensation with methyl bromopyruvate proceeds under mild reflux conditions in acetone, demonstrating excellent functional group tolerance. By avoiding complex protecting group strategies and minimizing the total number of isolation steps, this route significantly enhances the overall yield and purity of the final amide products. This efficiency translates directly into commercial advantages, allowing for cost reduction in electronic chemical manufacturing and pharmaceutical sectors alike by simplifying the operational workflow and reducing solvent consumption.
Mechanistic Insights into Phosphoric Acid-Catalyzed Cyclization
The cornerstone of this synthetic route is the initial formation of the substituted 2-aminonaphthyridine core, which sets the stage for all subsequent functionalization. As illustrated in the reaction scheme below, the condensation of 2,6-diaminopyridine with a 1,3-diketone derivative in the presence of phosphoric acid proceeds through a concerted mechanism that facilitates double bond formation and ring closure simultaneously. The acidic environment protonates the carbonyl oxygen of the diketone, increasing its electrophilicity and promoting nucleophilic attack by the amino groups of the pyridine ring. This cascade reaction is highly regioselective, ensuring that the nitrogen atoms are incorporated into the heterocyclic framework in the correct orientation to support biological activity. The use of phosphoric acid not only drives the equilibrium towards the product but also serves as a solvent medium that stabilizes the transition states, resulting in high conversion rates even at moderate temperatures around 90°C.
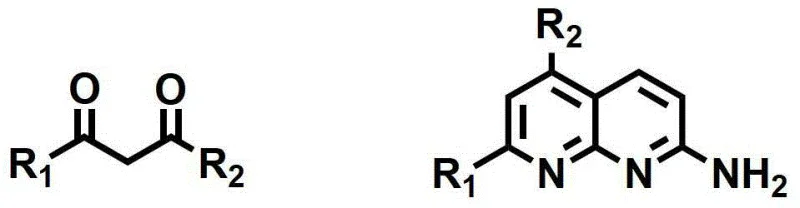
Following the core assembly, the introduction of the imidazole moiety is achieved through a nucleophilic substitution and cyclization sequence with methyl bromopyruvate. The amino group at the 2-position of the naphthyridine attacks the alpha-carbon of the bromopyruvate, displacing the bromide ion and forming a new carbon-nitrogen bond. Subsequent intramolecular cyclization closes the five-membered imidazole ring, creating the fused tricyclic system characteristic of the target compounds. The final transformation involves the activation of the carboxylic acid, generated via hydrolysis of the methyl ester, using HATU (2-(7-azobenzotriazole)-N,N,N',N'-tetramethyluronium hexafluorophosphate) and DIPEA. This coupling protocol is renowned for its ability to minimize racemization and suppress side reactions, ensuring that the diverse array of amine substituents can be attached with high fidelity. This mechanistic robustness is crucial for maintaining batch-to-batch consistency, a key requirement for any reliable agrochemical intermediate supplier or pharma partner aiming for regulatory approval.

How to Synthesize Imidazo[1,2-a][1,8]-naphthyridine Amides Efficiently
Implementing this synthesis on an industrial scale requires careful attention to reaction parameters and workup procedures to maximize yield and safety. The process begins with the precise stoichiometric mixing of the diamine and diketone in phosphoric acid, followed by controlled heating to ensure complete conversion without degradation. The subsequent steps involving esterification, hydrolysis, and amidation are all performed using standard unit operations familiar to process chemists, such as reflux, filtration, and liquid-liquid extraction. Detailed standardized synthesis steps see the guide below for specific molar ratios and temperature profiles that have been optimized to reduce impurity formation. By adhering to these protocols, manufacturers can achieve the high-purity OLED material or pharmaceutical intermediate standards necessary for downstream applications.
- Cyclization of 2,6-diaminopyridine with substituted diketones in phosphoric acid at 90°C to form the naphthyridine core.
- Condensation with methyl bromopyruvate in acetone under reflux to construct the imidazole ring.
- Hydrolysis of the methyl ester using lithium hydroxide to yield the carboxylic acid intermediate.
- Final amide coupling with various amines using HATU and DIPEA in DMF to generate the target compound.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic sourcing perspective, the adoption of this synthetic route offers profound benefits for supply chain resilience and cost management. The reliance on commodity chemicals rather than bespoke intermediates drastically reduces the risk of supply disruptions caused by single-source dependencies. Furthermore, the elimination of transition metal catalysts removes the need for expensive scavenging resins and extensive analytical testing for heavy metal residues, which are often major cost drivers in API production. This simplification of the purification train allows for faster turnaround times and lower capital expenditure on specialized equipment. Consequently, partners can expect substantial cost savings and enhanced flexibility in scaling production volumes to meet fluctuating market demands without compromising on quality or regulatory compliance standards.
- Cost Reduction in Manufacturing: The process utilizes inexpensive and widely available starting materials like 2,6-diaminopyridine and simple diketones, which are produced on a multi-ton scale globally, ensuring stable pricing and availability. By avoiding the use of precious metal catalysts such as palladium or platinum, the method eliminates the high costs associated with catalyst procurement, recovery, and the rigorous removal of trace metals from the final product. Additionally, the high efficiency of the HATU-mediated coupling step minimizes the loss of valuable intermediates, further driving down the cost of goods sold (COGS) for the final active pharmaceutical ingredient. This economic efficiency makes the technology particularly attractive for generic drug manufacturers seeking to optimize their margins while maintaining competitive pricing in the antiviral market.
- Enhanced Supply Chain Reliability: The synthetic pathway is designed around robust chemistry that tolerates minor variations in reaction conditions, making it highly suitable for transfer between different manufacturing sites or contract development and manufacturing organizations (CDMOs). Since the raw materials are not subject to complex geopolitical restrictions or limited production capacity, procurement teams can secure long-term supply agreements with greater confidence. The simplicity of the workup procedures, which primarily involve precipitation and filtration, reduces the dependency on specialized chromatography columns that can be bottlenecks in large-scale production. This operational robustness ensures a continuous flow of materials, mitigating the risk of stockouts that could delay clinical trials or commercial launches of life-saving HCV medications.
- Scalability and Environmental Compliance: The reaction conditions employed, such as the use of phosphoric acid and aqueous workups, align well with green chemistry principles by reducing the generation of hazardous organic waste streams. The process avoids the use of chlorinated solvents in the key cyclization step, favoring safer alternatives like acetone and DMF which are easier to recover and recycle. This environmental profile simplifies the permitting process for new manufacturing facilities and reduces the costs associated with waste disposal and treatment. Moreover, the high yields reported in the patent examples indicate that the process is inherently scalable, allowing for seamless transition from kilogram-level pilot batches to multi-ton commercial production without the need for significant re-optimization of reaction parameters.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented technology. These insights are derived directly from the experimental data and claims presented in the patent documentation, providing a clear understanding of the method's capabilities and limitations. Understanding these details is essential for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: What is the primary therapeutic application of these imidazonaphthyridine compounds?
A: According to patent CN103333168A, these compounds exhibit potent inhibitory activity against the Hepatitis C Virus (HCV), specifically targeting viral proliferation with IC50 values in the nanomolar range, offering a potential alternative to interferon-based therapies.
Q: Does this synthesis route require expensive transition metal catalysts?
A: No, the disclosed method relies on phosphoric acid for the initial cyclization and standard peptide coupling reagents like HATU for the final step, eliminating the need for costly palladium or other transition metal catalysts and simplifying purification.
Q: How does this method improve supply chain reliability for API production?
A: The process utilizes commercially available and inexpensive starting materials such as 2,6-diaminopyridine and simple diketones, reducing dependency on complex custom synthons and ensuring a more robust and continuous supply chain for large-scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Imidazo[1,2-a][1,8]-naphthyridine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of having a dependable partner who can navigate the complexities of synthesizing advanced heterocyclic intermediates. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project moves smoothly from benchtop discovery to full-scale manufacturing. We are committed to delivering high-purity intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Whether you require custom synthesis of specific analogs or bulk supply of the core scaffold described in CN103333168A, our infrastructure is designed to support your timelines and quality requirements with precision and reliability.
We invite you to engage with our technical procurement team to discuss how we can tailor our capabilities to your specific needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into how our optimized processes can reduce your overall project costs. We encourage potential partners to contact us for specific COA data and route feasibility assessments, allowing you to make informed decisions based on hard data and proven expertise. Let us help you accelerate your HCV inhibitor development program with our superior supply chain solutions and technical excellence.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →