Scalable Stereoselective Synthesis of Purine Dioxolane Nucleosides for Commercial API Production
The pharmaceutical industry's relentless pursuit of effective antiviral therapies has placed significant emphasis on the efficient production of nucleoside analogs, particularly those targeting HIV and HBV. Patent CN101801382B discloses a groundbreaking stereoselective process for preparing purine dioxolane nucleoside derivatives, specifically addressing the critical need for cost-effective and scalable manufacturing methods. This technology represents a paradigm shift from traditional multi-step asymmetric syntheses to a more direct, additive-promoted glycosylation strategy. By leveraging specific alpha-cyanocarbonyl compounds as reaction additives, the process achieves superior chemical yields and exceptional stereoselectivity, directly impacting the commercial viability of key antiviral intermediates like (-)-DAPD (Amdoxovir). For R&D directors and procurement specialists, understanding the mechanistic nuances of this patent is essential for securing a reliable supply chain for high-purity API intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of enantiomerically pure beta-D-1,3-dioxolane nucleosides has been plagued by significant technical hurdles that hinder commercial scalability. Early methodologies, such as the asymmetric synthesis reported by Chu and Shinazi, relied on starting materials like 1,6-dehydration-D-mannose and involved arduous 13-step sequences. These routes were characterized by low overall yields, difficult oxidation steps, and complex purification protocols that made them economically unfeasible for large-scale production. Furthermore, alternative approaches disclosed in documents like PCT WO9721706 attempted to utilize coupling reactions at extremely low temperatures, specifically around -78°C. While these cryogenic conditions offered some degree of stereoselectivity, they resulted in poor chemical yields and required reaction times exceeding 24 hours, creating substantial energy costs and operational bottlenecks. Another reported method using optional silylated 1,3-dicarbonyl compounds still suffered from limited utility, achieving only a 33% yield of the desired cis-isomer during the critical glycosylation step, which is insufficient for robust commercial development.
The Novel Approach
In stark contrast to these legacy methods, the process described in CN101801382B introduces a highly efficient pathway that bypasses the need for extreme cryogenic conditions and excessive synthetic steps. The core innovation lies in the direct reaction of a purine or polysilylated purine derivative with an activated dioxolane analog in the presence of a specific additive. This additive, existing in the form of an alpha-cyanocarbonyl compound or its silylated derivative, fundamentally alters the reaction landscape. By conducting the glycosylation at much milder temperatures, ranging from -25°C to the solvent boiling point (preferably -10°C to 30°C), the process drastically reduces energy consumption and equipment complexity. The result is a reaction that proceeds with the highest reported chemical yields and excellent stereoselectivity, effectively solving the yield/selectivity trade-off that has long plagued this chemical class. This novel approach transforms the production of purine dioxolane nucleoside derivatives from a laboratory curiosity into a commercially viable manufacturing reality.
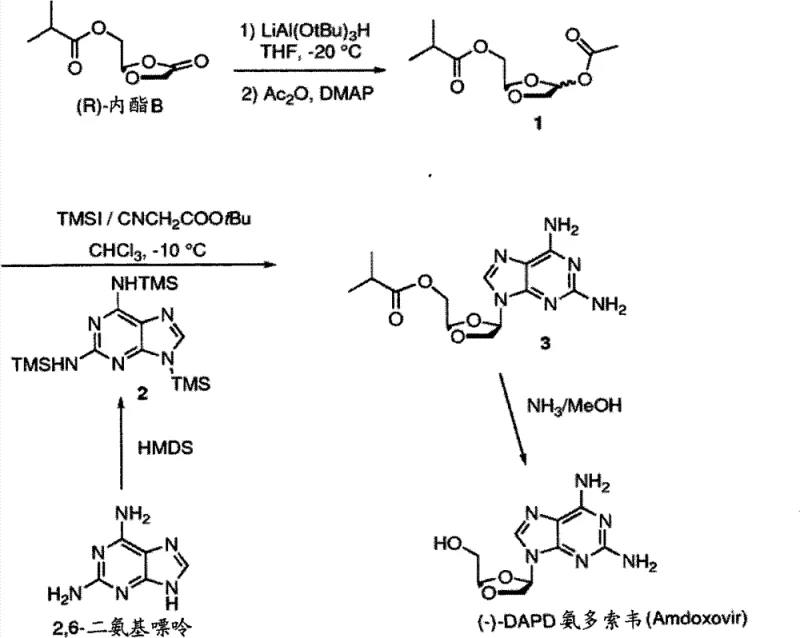
Mechanistic Insights into Additive-Promoted Stereoselective Glycosylation
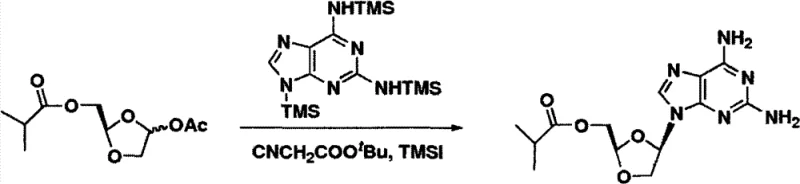
The success of this stereoselective process hinges on the intricate interplay between the Lewis acid catalyst, the activated sugar donor, and the crucial alpha-cyano carbonyl additive. The reaction initiates with the activation of the dioxolane acetate, typically generated from the reduction of a lactone precursor using bulky reducing agents like lithium tri-tert-butoxyaluminum hydride. Upon exposure to a Lewis acid such as trimethylsilyl iodide (TMSI), the acetate leaving group is displaced, generating a reactive oxocarbenium ion intermediate. In conventional systems without the additive, this intermediate is prone to non-selective attack by the purine base, leading to a mixture of alpha and beta anomers. However, the presence of the alpha-cyano carbonyl additive, such as tert-butyl cyanoacetate, appears to stabilize the transition state or coordinate with the Lewis acid in a manner that shields one face of the oxocarbenium ion. This steric and electronic modulation forces the nucleophilic attack of the silylated 2,6-diaminopurine to occur preferentially from the beta-face, thereby enriching the desired stereoisomer.
Impurity control is another critical aspect where this mechanism offers distinct advantages. In traditional glycosylations, the formation of the wrong anomer often necessitates difficult chromatographic separations that are impractical on a multi-ton scale. The additive-promoted mechanism described here shifts the product distribution significantly towards the target beta-isomer, as evidenced by the isomer ratios observed in the experimental data. Following the coupling reaction, the crude product can be purified through straightforward recrystallization techniques, often using simple solvents like isopropyl alcohol. This ability to upgrade purity through crystallization rather than chromatography is a hallmark of a robust industrial process. It ensures that the final API intermediate meets stringent purity specifications required by regulatory bodies, minimizing the risk of genotoxic impurities or isomeric contaminants that could compromise drug safety and efficacy.
How to Synthesize (-)-DAPD Efficiently
The synthesis of (-)-DAPD and related analogs via this patented route involves a logical sequence of protection, activation, coupling, and deprotection steps that are amenable to standard chemical engineering practices. The process begins with the silylation of the purine base to enhance its solubility and nucleophilicity, followed by the preparation of the activated sugar donor. The pivotal coupling step utilizes the aforementioned alpha-cyano additive to drive stereoselectivity, followed by a final deprotection stage to reveal the active hydroxyl groups. This streamlined workflow eliminates the need for chiral pool starting materials and reduces the total number of unit operations. For process chemists looking to implement this technology, the detailed standardized synthesis steps are outlined below to ensure reproducibility and safety.
- Silylate 2,6-diaminopurine using hexamethyldisilazane (HMDS) and ammonium sulfate to protect amino groups and enhance nucleophilicity.
- Activate the dioxolane sugar precursor by reducing the lactone with LiAl(OtBu)3H followed by acetylation to form the acetoxylated intermediate.
- Perform the glycosylation coupling reaction between the silylated purine and activated sugar in the presence of TMSI and a tert-butyl cyanoacetate additive to ensure high beta-selectivity.
- Remove the protecting groups via ammonolysis in methanol to yield the final (-)-DAPD nucleoside analog.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this stereoselective process offers tangible strategic benefits that extend beyond mere technical feasibility. The primary advantage lies in the drastic simplification of the manufacturing workflow, which directly correlates to reduced operational expenditures and enhanced supply reliability. By eliminating the need for complex asymmetric synthesis starting from carbohydrates like mannose, manufacturers can source simpler, more abundant raw materials. This shift reduces dependency on specialized chiral suppliers and mitigates the risk of raw material shortages that often plague the pharmaceutical supply chain. Furthermore, the ability to operate at near-ambient temperatures removes the requirement for specialized cryogenic reactors, allowing production to be conducted in standard glass-lined steel vessels available in most multipurpose chemical plants.
- Cost Reduction in Manufacturing: The economic impact of this process is driven by the elimination of expensive and energy-intensive processing conditions. Traditional methods requiring -78°C cooling demand significant refrigeration capacity and extended reaction times, both of which inflate utility costs. By shifting the reaction window to -10°C to 30°C, the process achieves substantial energy savings. Additionally, the improved stereoselectivity reduces the loss of valuable intermediates to unwanted isomers, effectively increasing the overall mass balance efficiency. The removal of transition metal catalysts or complex chiral auxiliaries further simplifies the waste stream, lowering the costs associated with hazardous waste disposal and environmental compliance.
- Enhanced Supply Chain Reliability: Supply continuity is paramount for life-saving antiviral medications. This synthetic route enhances reliability by utilizing robust, commodity-grade reagents such as hexamethyldisilazane, trimethylsilyl iodide, and common esters. These materials are widely available from multiple global suppliers, preventing single-source bottlenecks. The process tolerance is also improved; the reaction is less sensitive to minor fluctuations in temperature compared to cryogenic methods, ensuring consistent batch-to-batch quality. This robustness allows for tighter production scheduling and faster turnaround times, enabling manufacturers to respond more agilely to market demand surges without compromising product integrity.
- Scalability and Environmental Compliance: Scaling chemical processes from the bench to the plant floor often introduces unforeseen challenges, but this methodology is inherently designed for scale-up. The reliance on recrystallization for purification, rather than column chromatography, is a critical factor for industrial viability. Crystallization is a standard unit operation that scales linearly and produces high-purity solids with minimal solvent usage. This aligns with green chemistry principles by reducing solvent waste volumes. Moreover, the simplified workup procedures, involving basic aqueous washes and phase separations, facilitate easier handling of large volumes, ensuring that the process remains environmentally compliant and safe for operators even at multi-ton production levels.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this stereoselective synthesis technology. These insights are derived directly from the patent specifications and are intended to clarify the operational parameters and benefits for potential partners. Understanding these details is crucial for evaluating the feasibility of integrating this route into existing manufacturing portfolios.
Q: How does the alpha-cyano carbonyl additive improve stereoselectivity in this synthesis?
A: The addition of alpha-cyano carbonyl compounds, such as tert-butyl cyanoacetate, during the glycosylation step significantly enhances the beta/anomeric selectivity. This additive interacts with the Lewis acid catalyst and the oxocarbenium ion intermediate, favoring the formation of the desired cis-beta isomer over the alpha isomer, thereby reducing the burden on downstream purification.
Q: What are the advantages of this method over traditional asymmetric synthesis routes?
A: Traditional asymmetric syntheses often involve lengthy sequences, such as the 13-step route from 1,6-dehydration-D-mannose, which suffers from low overall yields and difficult oxidation steps. This patented process utilizes a direct coupling of activated dioxolane analogs with silylated purines, drastically shortening the synthetic sequence and avoiding complex chiral pool starting materials while maintaining high optical purity through crystallization.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the process is designed for scalability. It operates at moderate temperatures (-10°C to 30°C) rather than extreme cryogenic conditions (-78°C) required by previous methods. Furthermore, the use of recrystallization for purification, specifically using isopropyl alcohol, is a unit operation that translates efficiently from laboratory to multi-ton production scales, ensuring consistent quality and supply continuity.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable (-)-DAPD Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthesis routes for next-generation antiviral therapeutics. Our technical team has extensively analyzed the methodology disclosed in CN101801382B and possesses the expertise to translate this laboratory-scale innovation into commercial reality. We have extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. Our facility is equipped with rigorous QC labs and advanced analytical instrumentation to guarantee stringent purity specifications for all nucleoside intermediates, adhering to the highest international regulatory standards.
We invite you to collaborate with us to optimize your supply chain for purine dioxolane nucleoside derivatives. Our engineering team is ready to conduct a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. By leveraging our process development capabilities, we can help you navigate the complexities of scale-up and regulatory filing. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how our manufacturing excellence can drive value for your organization.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →