Advanced Organocatalytic Synthesis of Chiral 3-Hydroxy-3-Nitromethyl Oxindoles for Pharmaceutical Manufacturing
Introduction to Novel Asymmetric Synthesis Technology
The pharmaceutical industry continuously seeks robust methodologies for constructing quaternary carbon stereocenters, particularly within the privileged 3-substituted-3-hydroxy-2-oxindole scaffold found in numerous bioactive natural products and drug candidates. Patent CN102351777A discloses a groundbreaking preparation method for chiral 3-hydroxy-3-methylene nitroindol-2-one derivatives, utilizing isatin derivatives and nitromethane as primary substrates. This technology represents a significant leap forward in asymmetric organocatalysis, employing a specific cinchona alkaloid-derived catalyst to achieve high yields and excellent enantioselectivity under mild conditions. The ability to access these complex chiral building blocks efficiently is critical for the development of next-generation therapeutic agents targeting various diseases. By leveraging this patented approach, manufacturers can secure a reliable supply chain for high-purity pharmaceutical intermediates that were previously difficult to synthesize with such precision.
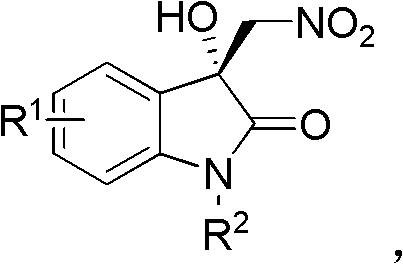
The core innovation lies in the direct asymmetric Henry reaction (nitroaldol reaction) between isatins and nitromethane, a transformation that constructs a challenging all-carbon quaternary stereocenter at the C3 position of the oxindole ring. Unlike traditional methods that might require harsh reagents or multi-step protection-deprotection sequences, this protocol streamlines the synthesis into a single catalytic step. The resulting nitro-oxindole products serve as versatile precursors; the nitro group can be readily transformed into amines, carboxylic acids, or other functional groups, thereby expanding the chemical space available for medicinal chemistry campaigns. For R&D directors evaluating new routes, this methodology offers a direct path to diverse libraries of chiral oxindoles, accelerating the lead optimization phase in drug discovery programs.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the asymmetric synthesis of 3-substituted-3-hydroxy-2-oxindoles has relied heavily on transition metal catalysis or stoichiometric chiral auxiliaries, which present significant drawbacks for industrial application. Many conventional protocols utilize expensive noble metals such as palladium, rhodium, or copper complexes, which not only inflate raw material costs but also introduce stringent regulatory hurdles regarding heavy metal residues in final API products. Furthermore, these metal-catalyzed reactions often require strictly anhydrous conditions, cryogenic temperatures below -78°C, or sensitive ligands that degrade upon exposure to air or moisture, complicating process scale-up. Alternative approaches involving aldol condensations with aldehydes or ketones often suffer from limited substrate scope or poor diastereoselectivity, necessitating difficult chromatographic separations that reduce overall throughput. These inefficiencies create bottlenecks in manufacturing, leading to longer lead times and higher production costs for complex pharmaceutical intermediates.
The Novel Approach
In stark contrast, the methodology described in CN102351777A utilizes a metal-free organocatalytic system that operates effectively in common organic solvents like tetrahydrofuran (THF) at a manageable -30°C. This novel approach employs a bifunctional cinchona alkaloid derivative, specifically a C9-benzyloxy-C6'-hydroxy-quinine derivative, which activates both the nucleophile (nitromethane) and the electrophile (isatin) simultaneously through hydrogen bonding networks. This dual activation mode ensures high stereocontrol without the need for toxic metals, inherently simplifying the workup procedure and eliminating the need for expensive metal scavengers. The reaction demonstrates remarkable tolerance to various substituents on the isatin ring, including halogens and alkyl groups, maintaining high yields (often exceeding 90%) and good enantiomeric excess (ee) values across a broad range of substrates. This robustness makes the process highly attractive for commercial scale-up, offering a sustainable and cost-effective alternative to legacy synthetic routes.
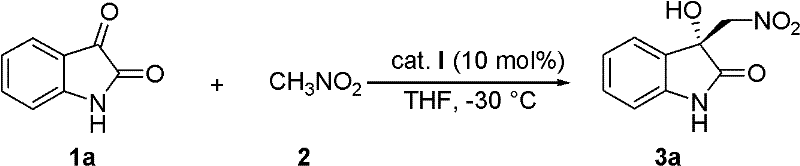
Mechanistic Insights into Cinchona Alkaloid-Catalyzed Henry Reaction
The success of this synthesis hinges on the precise molecular recognition and activation provided by the chiral catalyst, which dictates the stereochemical outcome of the C-C bond formation. The catalyst, structurally characterized as 4-((R)-(benzyloxy)((1S,2S,4S,5R)-5-vinylquinuclidin-2-yl)methyl)quinolin-6-ol, functions as a bifunctional organocatalyst. The tertiary amine moiety of the quinuclidine ring acts as a Brønsted base to deprotonate nitromethane, generating a reactive nitronate species, while the phenolic hydroxyl group and potentially the ether oxygen engage in hydrogen bonding with the carbonyl oxygen of the isatin substrate. This organized transition state rigidly holds the reactants in a specific orientation, shielding one face of the isatin carbonyl and directing the attack of the nitronate to the opposite face, thus establishing the absolute configuration of the newly formed quaternary center. Understanding this mechanistic nuance is vital for process chemists aiming to optimize reaction parameters or adapt the catalyst for analogous transformations.
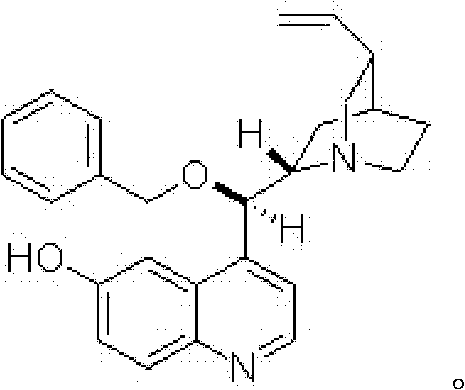
Furthermore, the choice of solvent and temperature plays a pivotal role in modulating the catalyst's conformational flexibility and the stability of the transition state assembly. The patent specifies THF as the preferred solvent, likely due to its ability to solubilize both the polar nitromethane and the organic substrates while supporting the necessary hydrogen-bonding interactions without competing excessively for them. The reaction temperature of -30°C strikes a balance between reaction rate and stereocontrol; lower temperatures generally favor higher enantioselectivity by minimizing background non-catalyzed reactions and tightening the transition state geometry. Impurity control is inherently managed by the high selectivity of the catalyst, which minimizes the formation of diastereomers or regioisomers that often plague less selective methods. This high level of purity directly translates to simplified downstream processing, as fewer impurities need to be removed before the intermediate can be used in subsequent coupling reactions.
How to Synthesize Chiral 3-Hydroxy-3-Nitromethyl Oxindoles Efficiently
Implementing this synthesis requires careful attention to reagent quality and reaction monitoring to ensure consistent results. The general procedure involves dissolving the specific isatin derivative and the chiral catalyst in dry THF, followed by the addition of excess nitromethane to drive the equilibrium towards product formation. The reaction mixture is then stirred at -30°C for a duration that varies from several hours to several days, depending on the electronic nature of the isatin substituents; electron-deficient isatins typically react faster than electron-rich ones. Upon completion, the reaction is quenched with water, and the product is extracted into an organic phase, washed, dried, and purified via standard silica gel chromatography. For detailed operational parameters, stoichiometry, and specific workup instructions tailored to different substrates, please refer to the standardized synthesis guide below.
- Dissolve the isatin derivative substrate and the cinchona alkaloid-derived chiral catalyst (10 mol%) in anhydrous tetrahydrofuran (THF) under inert atmosphere.
- Add nitromethane (10 equivalents) to the reaction mixture and maintain the temperature at -30°C for a period ranging from 1 hour to 5 days depending on the substrate.
- Quench the reaction with water, extract with ethyl acetate, wash with brine, dry over anhydrous sodium sulfate, and purify the crude product via flash column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this organocatalytic technology offers substantial strategic benefits that align with modern manufacturing goals of sustainability and cost efficiency. The elimination of transition metals removes a major cost driver associated with catalyst acquisition and, more importantly, the extensive purification steps required to meet regulatory limits for heavy metals in pharmaceuticals. This simplification of the downstream process significantly reduces the consumption of solvents and adsorbents, lowering the overall environmental footprint and waste disposal costs. Additionally, the starting materials—isatin derivatives and nitromethane—are commodity chemicals available from multiple global suppliers, ensuring a stable and competitive supply chain that is not reliant on single-source proprietary reagents. This raw material accessibility mitigates supply risk and allows for flexible sourcing strategies to optimize purchasing costs.
- Cost Reduction in Manufacturing: The absence of precious metals and the use of low catalyst loading drastically reduce the direct material costs associated with the catalytic system. Moreover, the mild reaction conditions (-30°C) are achievable with standard industrial cooling equipment, avoiding the extreme energy expenditures required for cryogenic processes below -78°C. The high yields reported in the patent minimize raw material waste, ensuring that a greater proportion of input costs are converted into valuable product. By streamlining the purification workflow and reducing the number of unit operations, manufacturers can achieve significant operational expenditure savings, making the final intermediate more price-competitive in the global market.
- Enhanced Supply Chain Reliability: The robustness of the reaction against moisture and air, typical of many organocatalytic systems compared to sensitive metal complexes, enhances process reliability and reduces the risk of batch failures due to environmental excursions. The broad substrate scope means that a single catalytic platform can be used to produce a wide variety of analogues, allowing manufacturers to respond quickly to changing demand for different API intermediates without retooling or developing entirely new processes. This flexibility supports a more agile supply chain capable of adapting to the dynamic needs of pharmaceutical clients, ensuring continuity of supply even when specific precursor availability fluctuates.
- Scalability and Environmental Compliance: Scaling organocatalytic reactions is generally more straightforward than metal-catalyzed ones because there is no need to manage metal leaching or catalyst recovery systems designed for trace metal removal. The use of THF and nitromethane, while requiring standard safety protocols, fits well within existing solvent recovery infrastructures in chemical plants. The process generates minimal hazardous waste compared to stoichiometric chiral auxiliary methods, aligning with green chemistry principles and increasingly strict environmental regulations. This compliance ease facilitates faster regulatory approval for manufacturing sites and reduces the long-term liability associated with hazardous waste management.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. They are derived from the specific experimental data and beneficial effects outlined in the patent documentation, providing clarity on performance metrics and operational feasibility. Understanding these details helps stakeholders assess the suitability of this method for their specific production requirements and quality standards.
Q: What are the key advantages of this organocatalytic method over traditional metal-catalyzed routes?
A: This method eliminates the need for expensive and toxic transition metal catalysts, simplifying downstream purification and reducing heavy metal residue risks in pharmaceutical intermediates. It operates under mild conditions (-30°C) with high enantioselectivity.
Q: What is the scope of substrates compatible with this synthesis protocol?
A: The protocol demonstrates broad substrate tolerance, successfully converting various isatin derivatives including those with electron-withdrawing groups (Cl, Br, F) and electron-donating groups (Me), as well as N-substituted isatins (N-Me, N-Bn).
Q: How does the catalyst loading impact the economic feasibility of the process?
A: The process utilizes a relatively low catalyst loading of 10 mol%, which balances reaction efficiency with cost. The use of commercially available cinchona alkaloid derivatives further enhances the economic viability for large-scale production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral 3-Hydroxy-3-Nitromethyl Oxindole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-quality chiral intermediates in the development of life-saving medications. Our technical team has thoroughly analyzed the potential of the organocatalytic route described in CN102351777A and is prepared to leverage this technology for your projects. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. Our facilities are equipped with rigorous QC labs and advanced analytical instrumentation to guarantee stringent purity specifications, including precise control over enantiomeric excess and impurity profiles, meeting the exacting standards of the global pharmaceutical industry.
We invite you to collaborate with us to optimize this synthesis for your specific needs, whether for clinical trial material or commercial manufacturing. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are ready to provide specific COA data for similar compounds and conduct comprehensive route feasibility assessments to demonstrate how our expertise can accelerate your project timelines and reduce your overall cost of goods sold.