Advanced One-Step Synthesis of Pyrazolo[4,3-c]quinoline Derivatives for Commercial Scale-up
The pharmaceutical and fine chemical industries are constantly seeking more efficient pathways to access bioactive heterocyclic scaffolds, particularly those with proven therapeutic potential. Patent CN114213412B introduces a groundbreaking methodology for the synthesis of pyrazolo[4,3-c]quinoline derivatives, a class of fused heterocyclic compounds known for their diverse pharmacological activities including anti-cancer, anti-inflammatory, and antioxidant properties. These compounds have historically shown specific physiological effects on benzodiazepine receptors and act as selective cyclooxygenase-2 inhibitors, making them highly valuable targets for drug discovery programs. The disclosed innovation utilizes a novel acid-catalyzed strategy where 2-pyrazoleaniline derivatives react directly with ether compounds in a single step. This approach represents a significant departure from conventional multi-step syntheses, offering a streamlined route that leverages trifluoromethanesulfonic acid to facilitate ether bond cleavage and subsequent ring construction. By providing a robust platform for generating these complex structures, this technology addresses critical bottlenecks in the production of high-purity pharmaceutical intermediates.
![General chemical structure of pyrazolo[4,3-c]quinoline derivatives showing variable substituents R1, R2, and R3'](/insights/img/pyrazoloquinoline-synthesis-acid-catalysis-pharma-supplier-20260306111847-01.png)
Historically, the construction of the pyrazolo[4,3-c]quinoline core has been a challenging endeavor for process chemists, often requiring laborious sequences that hinder rapid scale-up. Prior art, such as that described in patent CN200410004444.9, typically relies on constructing either the quinoline or pyrazole ring first, followed by a fusion step that frequently involves harsh reagents like phosphorus oxychloride. These traditional methods not only generate significant hazardous waste but also suffer from moderate yields and complex purification requirements due to the formation of multiple byproducts. Furthermore, the reliance on chlorinating agents necessitates rigorous corrosion-resistant equipment and specialized waste treatment protocols, driving up operational costs. In contrast, the novel approach detailed in the present patent data utilizes a direct cyclization strategy that bypasses these inefficiencies entirely. By employing readily available ether solvents as both the reaction medium and the carbon source for ring closure, the new method drastically simplifies the synthetic workflow. This shift from a multi-step, hazard-intensive process to a concise, one-pot transformation underscores a major technological leap in the manufacturing of these specialized heterocycles.
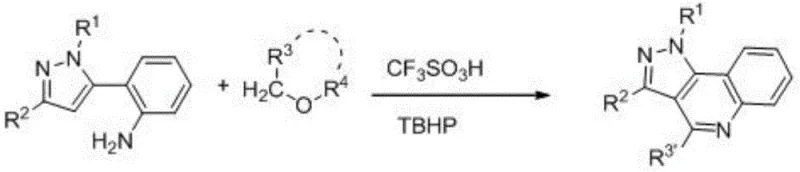
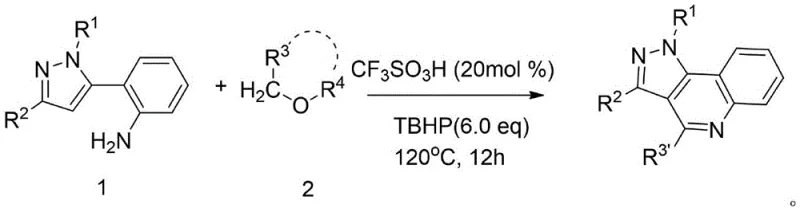
The mechanistic elegance of this synthesis lies in the activation of the ether substrate by a strong Brønsted acid, specifically trifluoromethanesulfonic acid (TfOH), which serves as a highly efficient catalyst without introducing metal contaminants. Under the reaction conditions, typically maintained at 120°C, the acid facilitates the cleavage of the ether bond, generating a reactive electrophilic species that attacks the electron-rich 2-pyrazoleaniline skeleton. This is accompanied by the use of tert-butyl hydroperoxide (TBHP) as an oxidant, which plays a crucial role in the aromatization of the newly formed quinoline ring. The absence of transition metal catalysts is a distinct advantage for pharmaceutical applications, as it eliminates the risk of heavy metal residues in the final active pharmaceutical ingredient (API), thereby reducing the burden on downstream purification processes. Moreover, the reaction demonstrates remarkable tolerance to various functional groups on the pyrazole ring, allowing for the incorporation of electron-withdrawing or electron-donating substituents without compromising the integrity of the cyclization. This mechanistic robustness ensures consistent product quality and minimizes the formation of difficult-to-remove impurities, which is a critical factor for maintaining stringent purity specifications in commercial production.
Mechanistic Insights into Triflic Acid-Catalyzed Cyclization
The core of this transformative process is the ability of trifluoromethanesulfonic acid to activate stable ether linkages, a feat that weaker acids often fail to achieve efficiently. In the catalytic cycle, the protonation of the ether oxygen increases the electrophilicity of the adjacent carbon atoms, rendering them susceptible to nucleophilic attack by the amino group of the 2-pyrazoleaniline precursor. This initial intermolecular interaction sets the stage for an intramolecular cyclization that constructs the quinoline moiety fused to the existing pyrazole ring. The presence of TBHP is essential for the final oxidative dehydrogenation step, ensuring the formation of the fully aromatic pyrazolo[4,3-c]quinoline system rather than a partially saturated intermediate. From an impurity control perspective, the mild nature of this acid-catalyzed environment compared to harsh chlorinating agents means fewer side reactions such as over-chlorination or polymerization occur. This results in a cleaner reaction profile, which is evidenced by the high isolated yields reported across various examples, ranging significantly above typical benchmarks for such complex heterocycle formations. The ability to tune the reaction by selecting different ethers—whether cyclic like tetrahydrofuran or acyclic like dibutyl ether—provides chemists with a versatile toolbox for introducing specific alkyl or hydroxyalkyl chains at the C4 position of the quinoline ring.
How to Synthesize Pyrazolo[4,3-c]quinoline Derivatives Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires careful attention to the stoichiometry of the oxidant and the choice of solvent, which doubles as a reactant. The procedure generally involves charging a sealed reaction vessel with the 2-pyrazoleaniline starting material, a catalytic amount of trifluoromethanesulfonic acid, and an excess of the selected ether compound. The addition of an aqueous solution of TBHP is then performed, and the mixture is heated to reflux conditions, typically around 120°C, for a duration of approximately 12 hours to ensure complete conversion. Following the reaction period, the workup procedure is straightforward, involving the removal of volatiles under reduced pressure followed by standard silica gel column chromatography to isolate the pure product. For detailed operational parameters and safety considerations regarding the handling of strong acids and peroxides, please refer to the standardized protocols below.
- Combine 2-pyrazoleaniline derivative, trifluoromethanesulfonic acid catalyst, and TBHP oxidant in a sealed tube with the chosen ether solvent.
- Heat the reaction mixture to 120°C and maintain stirring for approximately 12 hours to ensure complete cyclization.
- Concentrate the mixture under reduced pressure and purify the crude product via silica gel column chromatography to isolate the target compound.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic sourcing and manufacturing perspective, this patented methodology offers substantial benefits that directly impact the bottom line and supply chain resilience. The elimination of expensive and scarce transition metal catalysts removes a significant cost driver from the bill of materials, while also simplifying the regulatory dossier required for drug approval by avoiding heavy metal testing. The use of commodity chemicals such as tetrahydrofuran, dioxane, or simple alkyl ethers as reactants ensures a stable and reliable supply of raw materials, mitigating the risks associated with sourcing specialized reagents. Furthermore, the one-pot nature of the reaction reduces the number of unit operations required, leading to lower energy consumption and reduced labor costs per kilogram of product. The high yields observed across a broad range of substrates indicate a robust process that is less prone to batch-to-batch variability, enhancing overall production efficiency. These factors collectively contribute to a more sustainable and cost-effective manufacturing model for high-value pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The process achieves significant cost optimization by replacing costly metal catalysts with an inexpensive organic acid and utilizing bulk solvents as reactants. This substitution not only lowers the direct material costs but also reduces the expense associated with metal scavenging and waste disposal. The simplified workup procedure further decreases processing time and resource utilization, resulting in a leaner production workflow that maximizes throughput without compromising quality standards.
- Enhanced Supply Chain Reliability: By relying on widely available industrial chemicals like triflic acid and common ethers, the supply chain becomes less vulnerable to disruptions caused by the scarcity of specialized reagents. The robustness of the reaction conditions allows for flexible sourcing strategies, enabling procurement teams to negotiate better terms with multiple suppliers. Additionally, the stability of the intermediates and the final products ensures longer shelf life and easier logistics management, reducing the risk of inventory loss during transportation and storage.
- Scalability and Environmental Compliance: The absence of toxic phosphorus-based reagents and heavy metals aligns perfectly with modern green chemistry principles and stringent environmental regulations. This makes the process highly scalable from gram-scale research to multi-ton commercial production without the need for extensive retrofitting of waste treatment facilities. The reduced generation of hazardous byproducts simplifies effluent management, lowering the environmental footprint and ensuring compliance with increasingly strict global sustainability mandates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects outlined in the patent documentation, providing clarity on the practical aspects of adopting this method for large-scale production. Understanding these details is crucial for R&D directors and process engineers evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: What are the primary advantages of this acid-catalyzed method over traditional synthesis?
A: This method eliminates the need for toxic phosphorus oxychloride and complex multi-step sequences, offering a direct one-pot transformation with significantly higher yields and simpler purification.
Q: Does this process involve heavy metal catalysts that require removal?
A: No, the process utilizes trifluoromethanesulfonic acid as an organocatalyst, completely avoiding transition metals like palladium or copper, which simplifies regulatory compliance for pharmaceutical applications.
Q: What types of ether substrates are compatible with this synthesis route?
A: The method demonstrates broad substrate scope, successfully utilizing cyclic ethers like tetrahydrofuran and dioxane, as well as acyclic ethers such as dibutyl ether and benzyl methyl ether.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Pyrazolo[4,3-c]quinoline Supplier
As a leader in the custom synthesis of complex pharmaceutical intermediates, NINGBO INNO PHARMCHEM is uniquely positioned to leverage this advanced acid-catalyzed technology for your project needs. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial manufacturing is seamless and efficient. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of pyrazolo[4,3-c]quinoline derivatives meets the highest quality standards required by the global pharmaceutical industry. Our commitment to excellence extends beyond mere production; we act as a strategic partner dedicated to optimizing your supply chain and accelerating your time to market.
We invite you to engage with our technical procurement team to discuss how this innovative synthesis route can be tailored to your specific requirements. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits this process offers for your specific application. We encourage potential partners to reach out for specific COA data and route feasibility assessments, allowing us to demonstrate our capability to deliver high-purity intermediates with unmatched reliability and speed. Let us collaborate to bring your next generation of therapeutic agents from concept to reality.