Scalable Synthesis of High-Purity 4-Amino-Pyrimidine Derivatives for Targeted Cancer Therapy
Introduction to Patented Tyrosine Kinase Inhibitors
The pharmaceutical landscape is continuously evolving with the discovery of novel small molecules capable of interrupting critical cellular signaling pathways, and patent CN1212695A stands as a testament to this innovation by disclosing a comprehensive series of 4-amino-pyrimidine derivatives. These compounds are specifically engineered to inhibit signal transduction induced by tyrosine kinases, a mechanism that is pivotal in the progression of various malignancies and proliferative diseases. The patent outlines a robust chemical framework where the core pyrimidine structure is substituted with diverse amino groups, allowing for fine-tuning of pharmacological properties such as solubility, bioavailability, and receptor binding affinity. For R&D directors and procurement specialists, understanding the depth of this intellectual property is crucial, as it represents a verified pathway to developing next-generation oncology therapeutics. The versatility of the general formula described allows for the creation of a wide library of analogs, ensuring that drug discovery teams have ample chemical space to explore for optimal therapeutic indices while maintaining a clear freedom-to-operate landscape.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for constructing complex pyrimidine scaffolds often suffer from significant drawbacks that hinder their transition from bench-scale discovery to commercial manufacturing. Conventional methods frequently rely on harsh reaction conditions, such as extremely high temperatures or the use of toxic heavy metal catalysts, which introduce substantial challenges in terms of environmental compliance and worker safety. Furthermore, older methodologies often struggle with regioselectivity, leading to complex mixtures of isomers that require extensive and costly purification steps to isolate the desired active pharmaceutical ingredient. The reliance on exotic reagents or unstable intermediates in legacy processes can also create supply chain bottlenecks, making it difficult to secure consistent raw material quality. These inefficiencies not only drive up the cost of goods sold but also extend the lead time required to bring a new drug candidate to clinical trials, thereby delaying potential life-saving treatments for patients suffering from aggressive forms of cancer.
The Novel Approach
In contrast, the approach detailed in the provided patent data offers a streamlined and highly adaptable synthetic strategy that directly addresses the inefficiencies of legacy methods. By utilizing a nucleophilic aromatic substitution reaction between a halogenated pyrimidine precursor and a variety of amines, the process achieves high conversion rates under relatively mild conditions. This novel approach allows for the modular introduction of different side chains at specific positions on the pyrimidine ring, enabling rapid structure-activity relationship studies without the need to redesign the entire synthetic route. The use of common organic solvents such as isopropanol, toluene, or dimethylformamide ensures that the process is easily transferable to standard reactor equipment found in most contract development and manufacturing organizations. This flexibility significantly reduces the technical risk associated with scale-up, providing a clear path from milligram-scale synthesis to multi-ton commercial production while maintaining strict control over product quality and impurity profiles.
Mechanistic Insights into Nucleophilic Aromatic Substitution
The core chemical transformation described in this technology involves the reaction of a 4-amino-pyrimidine derivative bearing a leaving group, designated as Formula II, with an amine nucleophile, designated as Formula III. This nucleophilic aromatic substitution is driven by the electron-deficient nature of the pyrimidine ring, which activates the carbon-halogen bond towards attack by the nitrogen lone pair of the amine. The reaction mechanism proceeds through a Meisenheimer-like complex intermediate, where the stability of this intermediate is influenced by the electronic properties of the substituents on the pyrimidine ring and the nature of the leaving group. Understanding this mechanistic detail is vital for process chemists, as it allows for the rational selection of reaction parameters such as temperature and base strength to maximize yield and minimize side reactions. The patent specifies that leaving groups can include halogens like fluorine, chlorine, or bromine, as well as substituted hydroxyl or sulfonyl groups, providing a wide range of options for optimizing reaction kinetics and thermodynamics.
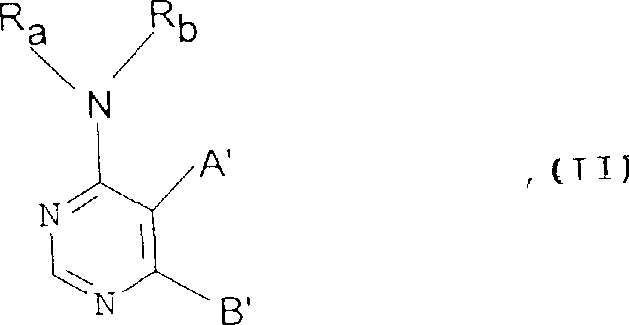
Controlling the impurity profile during this substitution reaction is paramount for ensuring the safety and efficacy of the final pharmaceutical product. The patent highlights that the reaction can be conducted in the presence of inorganic or organic bases, such as sodium carbonate, potassium hydroxide, or triethylamine, which serve to neutralize the acid byproduct generated during the substitution. Careful control of the stoichiometry and the addition rate of the amine nucleophile is essential to prevent over-alkylation or the formation of bis-substituted byproducts. Furthermore, the ability to conduct the reaction in a variety of solvents allows for the optimization of solubility parameters, ensuring that both reactants and products remain in solution or precipitate out as desired for easy isolation. This level of mechanistic control ensures that the resulting 4-amino-pyrimidine derivatives meet the stringent purity specifications required for clinical development, reducing the burden on downstream purification processes and enhancing the overall efficiency of the manufacturing workflow.
How to Synthesize 4-Amino-Pyrimidine Derivatives Efficiently
Executing the synthesis of these high-value intermediates requires a precise understanding of the reaction conditions and workup procedures outlined in the patent documentation. The process begins with the preparation of the halogenated pyrimidine precursor, which must be of high purity to ensure consistent reaction outcomes. Following this, the nucleophilic substitution is carried out in a suitable solvent system, with temperature control being a critical parameter to balance reaction rate and selectivity. The detailed standardized synthesis steps see the guide below, which provides a structured framework for replicating the experimental results on a larger scale. Adhering to these protocols ensures that the critical quality attributes of the intermediate are maintained, facilitating a smooth transition into subsequent drug substance manufacturing stages.
- Prepare the 4-amino-pyrimidine precursor (Formula II) containing a suitable leaving group such as fluorine or chlorine at the reactive position.
- React Formula II with the desired amine (Formula III) in a solvent like isopropanol or toluene, optionally with a base such as triethylamine or potassium carbonate.
- Purify the resulting crude mixture using chromatography or recrystallization to achieve high-purity pharmaceutical intermediates suitable for biological testing.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthetic route offers substantial benefits for procurement managers and supply chain heads looking to optimize their sourcing strategies for oncology intermediates. The reliance on readily available starting materials and common solvents significantly reduces the risk of supply disruptions, ensuring a continuous flow of materials necessary for uninterrupted production schedules. By eliminating the need for exotic catalysts or specialized equipment, the process lowers the barrier to entry for manufacturing partners, fostering a more competitive supplier landscape that drives down overall costs. This accessibility translates into enhanced supply chain reliability, allowing pharmaceutical companies to secure long-term agreements with confidence. Furthermore, the robustness of the chemistry means that yield losses due to process variability are minimized, contributing to a more predictable and efficient production cycle that aligns with just-in-time manufacturing principles.
- Cost Reduction in Manufacturing: The synthetic pathway described eliminates the need for expensive transition metal catalysts and complex purification steps, leading to significant cost savings in the overall manufacturing budget. By utilizing commodity chemicals and solvents that are available in bulk quantities, the process reduces raw material expenses and minimizes waste disposal costs associated with hazardous reagents. The high selectivity of the reaction reduces the formation of difficult-to-remove impurities, thereby lowering the consumption of chromatography media and solvents during the purification phase. These cumulative efficiencies result in a lower cost of goods sold, allowing for more competitive pricing of the final drug product while maintaining healthy profit margins for all stakeholders involved in the supply chain.
- Enhanced Supply Chain Reliability: The use of stable and commercially available reagents ensures that the supply chain is resilient against market fluctuations and geopolitical disruptions. Unlike processes that rely on single-source specialty chemicals, this method allows for multi-sourcing of key raw materials, reducing the risk of bottlenecks that can delay production timelines. The flexibility in solvent selection further enhances reliability, as manufacturers can switch between approved solvents based on availability without compromising product quality. This adaptability is crucial for maintaining continuous supply to clinical and commercial markets, ensuring that patients have uninterrupted access to vital medications. The robust nature of the process also simplifies technology transfer between sites, enabling rapid capacity expansion in response to increased demand.
- Scalability and Environmental Compliance: The reaction conditions are well-suited for large-scale production, operating within a temperature range that is easily manageable in standard industrial reactors without requiring extreme heating or cooling capabilities. The absence of heavy metals and the use of greener solvent options align with modern environmental, health, and safety standards, reducing the regulatory burden associated with waste management and emissions. This compliance facilitates faster regulatory approvals and reduces the risk of production shutdowns due to environmental violations. The scalability of the process ensures that it can meet the growing demand for oncology therapeutics without the need for significant capital investment in new infrastructure. This makes it an attractive option for contract manufacturing organizations looking to expand their portfolio of high-value pharmaceutical intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of these 4-amino-pyrimidine derivatives, based on the detailed specifications provided in the patent literature. These answers are designed to clarify the feasibility of the process and its alignment with industry standards for pharmaceutical development. Understanding these aspects helps stakeholders make informed decisions about integrating this technology into their existing pipelines. The information provided reflects the current state of the art as described in the intellectual property documentation.
Q: What is the primary biological mechanism of these 4-amino-pyrimidine derivatives?
A: These compounds function as potent inhibitors of signal transduction induced by tyrosine kinases, specifically targeting the Epidermal Growth Factor Receptor (EGF-R) pathway to suppress tumor growth and proliferation.
Q: Are the synthesis conditions compatible with large-scale manufacturing?
A: Yes, the patented process utilizes common organic solvents like isopropanol and toluene and operates within a flexible temperature range of 0 to 180 degrees Celsius, facilitating easy commercial scale-up.
Q: How is impurity control managed during the nucleophilic substitution?
A: Impurity control is achieved by selecting specific leaving groups such as halogens and optimizing reaction temperatures between 20 and 150 degrees Celsius, ensuring high selectivity and minimal byproduct formation.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 4-Amino-Pyrimidine Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of having a dependable partner for the production of complex pharmaceutical intermediates like the 4-amino-pyrimidine derivatives described in patent CN1212695A. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can move seamlessly from clinical trials to full-scale market launch. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch meets the highest quality standards required for oncology drug development. Our commitment to technical excellence means that we can navigate the complexities of nucleophilic substitution chemistry with precision, delivering materials that are ready for the next stage of your value chain.
We invite you to collaborate with us to optimize your supply chain and reduce your overall development costs through our Customized Cost-Saving Analysis. Our technical procurement team is ready to provide specific COA data and route feasibility assessments tailored to your unique project requirements. By leveraging our expertise, you can accelerate your time to market and ensure a stable supply of high-quality intermediates. Contact us today to discuss how we can support your oncology pipeline with reliable and scalable manufacturing solutions.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →