Advanced Synthesis of Enantiomerically Enriched Tetrahydrobenzothiepine Oxides for Cardiovascular Therapeutics
Introduction to Patent CN1284953A and Therapeutic Potential
The pharmaceutical landscape for treating cardiovascular disorders, particularly hypercholesterolemia and atherosclerosis, has seen significant advancements through the development of Ileal Bile Acid Transporter (IBAT) inhibitors. Patent CN1284953A discloses a groundbreaking process for the preparation of enantiomerically enriched tetrahydrobenzothiepine oxides, which serve as critical intermediates and active pharmaceutical ingredients in this therapeutic class. The core innovation lies in the ability to construct the complex seven-membered benzothiepine ring system with precise stereochemical control at the C4 and C5 positions, directly from chiral sulfoxide precursors. This methodology bypasses the traditional bottlenecks of racemic synthesis followed by difficult resolution, offering a more direct and efficient route to high-purity drug substances.
The structural versatility of these compounds is exemplified by Formula I, which allows for extensive substitution patterns on the phenyl ring and the seven-membered heterocycle. 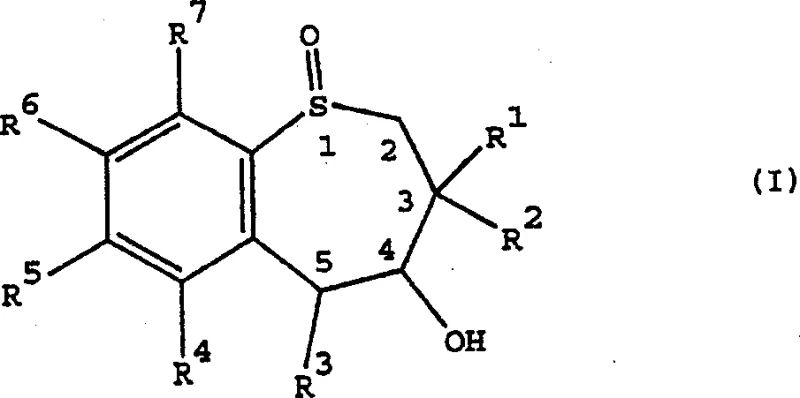 As depicted in the general structure, the molecule features a fused benzene and seven-membered sulfur-containing ring, where the sulfur atom itself can act as a chiral center or be oxidized to a sulfone. The ability to introduce diverse functional groups, such as dimethylamino or alkoxy substituents, enables the fine-tuning of pharmacokinetic properties essential for effective IBAT inhibition. For procurement teams seeking a reliable pharmaceutical intermediates supplier, understanding the robustness of this synthetic pathway is crucial for securing long-term supply chains for next-generation lipid-lowering therapies.
As depicted in the general structure, the molecule features a fused benzene and seven-membered sulfur-containing ring, where the sulfur atom itself can act as a chiral center or be oxidized to a sulfone. The ability to introduce diverse functional groups, such as dimethylamino or alkoxy substituents, enables the fine-tuning of pharmacokinetic properties essential for effective IBAT inhibition. For procurement teams seeking a reliable pharmaceutical intermediates supplier, understanding the robustness of this synthetic pathway is crucial for securing long-term supply chains for next-generation lipid-lowering therapies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methods for synthesizing benzothiazepine and related seven-membered ring systems have been plagued by significant inefficiencies that hinder commercial viability. For instance, earlier approaches described by Higashikawa relied on the optical resolution of racemic mixtures using chiral crown ether chromatography. While effective on a small scale, chromatographic separation is notoriously expensive and difficult to scale, leading to substantial material loss and high solvent consumption. Similarly, methods reported by Giordano required the use of pre-enriched amino acid starting materials, which limits the structural diversity of the final product and increases raw material costs due to the premium price of chiral pool resources.
Furthermore, biological approaches, such as the microbial reduction of diketones reported by Patel, introduce a different set of supply chain vulnerabilities. These processes are inherently limited by the capacity of bacterial cultures and often yield only microgram quantities, making them unsuitable for the kilogram-to-ton scale required for clinical and commercial drug manufacturing. The need to maintain specific microbial strains and control fermentation parameters adds layers of complexity and risk to the production process. Additionally, chemical reduction methods using sodium borohydride often result in racemic products that require further downstream processing, thereby increasing the overall step count and reducing the cumulative yield of the synthesis.
The Novel Approach
The process disclosed in CN1284953A represents a paradigm shift by utilizing an intramolecular cyclization of an enantiomerically enriched aryl-3-propionaldehyde sulfoxide. This strategy leverages the chirality already present at the sulfur atom to induce stereochemistry at the carbon atoms of the newly formed ring. By starting with a chiral sulfoxide, which can be prepared via highly selective oxidation of a sulfide, the synthesis effectively transfers chirality from the sulfur to the carbon backbone. This eliminates the need for resolving racemates post-cyclization, significantly improving the overall atom economy and process efficiency.
This novel approach also offers superior flexibility in terms of substrate scope. The method tolerates a wide range of substituents on the aromatic ring and the side chains, allowing for the synthesis of diverse analogues like the potent IBAT inhibitors shown in Formula (4R,5R)-XXVII. 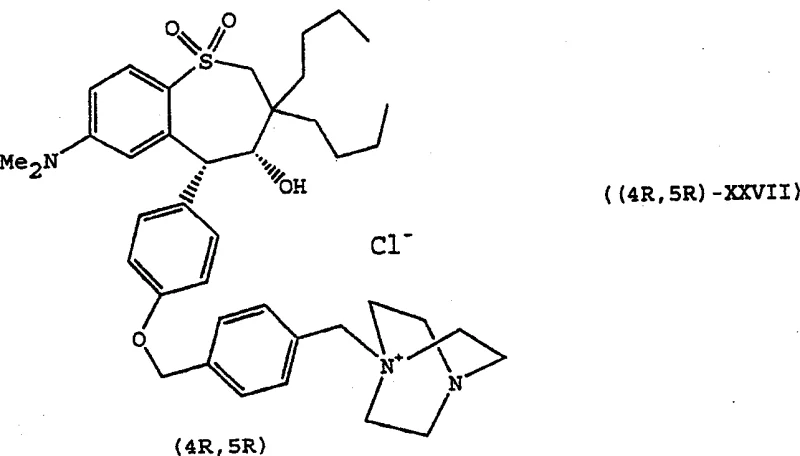 The use of standard chemical reagents such as potassium tert-butoxide for the cyclization step ensures that the reaction can be performed in common industrial solvents like THF under controlled temperatures. This chemical robustness translates directly into cost reduction in pharmaceutical intermediates manufacturing, as it avoids the specialized equipment and stringent controls required for bioprocessing or preparative chiral HPLC.
The use of standard chemical reagents such as potassium tert-butoxide for the cyclization step ensures that the reaction can be performed in common industrial solvents like THF under controlled temperatures. This chemical robustness translates directly into cost reduction in pharmaceutical intermediates manufacturing, as it avoids the specialized equipment and stringent controls required for bioprocessing or preparative chiral HPLC.
Mechanistic Insights into Base-Catalyzed Stereoselective Cyclization
The heart of this technology is the cyclization mechanism that converts the linear aryl-3-propionaldehyde sulfoxide (Formula II) into the cyclic tetrahydrobenzothiepine 1-oxide (Formula I). 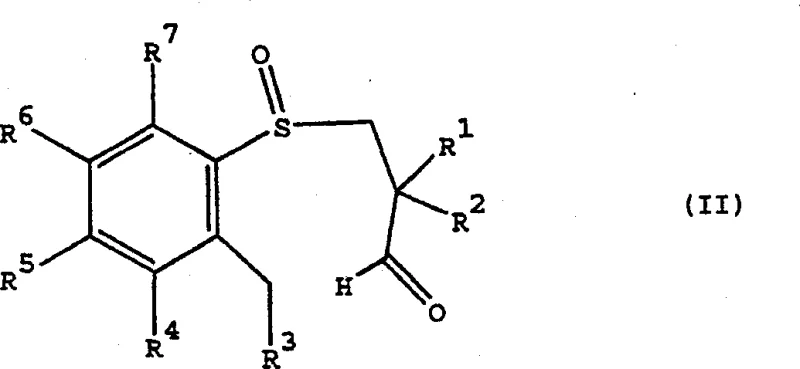 In this transformation, the aldehyde group at the end of the propyl chain becomes electrophilic, while the ortho-position of the aromatic ring acts as the nucleophile, facilitated by the base. However, the critical aspect is the stereochemical outcome. The sulfur atom, bearing a lone pair and an oxygen atom, creates a chiral environment that dictates the approach of the nucleophile. The transition state is organized such that the bulky substituents on the sulfur and the adjacent carbon chain minimize steric clash, leading to the preferential formation of either the (4R,5R) or (4S,5S) diastereomer depending on the configuration of the starting sulfoxide.
In this transformation, the aldehyde group at the end of the propyl chain becomes electrophilic, while the ortho-position of the aromatic ring acts as the nucleophile, facilitated by the base. However, the critical aspect is the stereochemical outcome. The sulfur atom, bearing a lone pair and an oxygen atom, creates a chiral environment that dictates the approach of the nucleophile. The transition state is organized such that the bulky substituents on the sulfur and the adjacent carbon chain minimize steric clash, leading to the preferential formation of either the (4R,5R) or (4S,5S) diastereomer depending on the configuration of the starting sulfoxide.
Impurity control is inherently built into this mechanism. Because the stereochemistry is determined during the bond-forming step rather than by separating isomers afterwards, the potential for generating the wrong enantiomer is minimized at the source. The patent highlights that selective reaction conditions can predominantly generate one specific isomer, such as the (4R,5R) compound, with high enantiomeric excess. This high level of purity is vital for R&D directors concerned with the safety profile of cardiovascular drugs, where the wrong enantiomer could exhibit off-target effects or toxicity. The subsequent oxidation of the sulfide to the sulfoxide using chiral oxidants like camphor-sulfonyl oxaziridines ensures that the input material for the cyclization is already highly enriched, further driving the purity of the final cyclic product.
How to Synthesize Tetrahydrobenzothiepine Oxides Efficiently
The synthesis of these complex heterocycles involves a logical sequence of oxidation and cyclization steps that are amenable to standard chemical manufacturing practices. The process begins with the preparation of an aryl-3-hydroxypropyl sulfide, which is then subjected to enantioselective oxidation to establish the first chiral center at the sulfur atom. Following this, the hydroxyl group is oxidized to an aldehyde, setting the stage for the ring-closing reaction. The final cyclization is triggered by a base, locking in the stereochemistry at the C4 and C5 positions. Detailed standardized synthesis steps for implementing this route are provided in the guide below.
- Perform enantioselective oxidation of an aryl-3-hydroxypropyl sulfide using a chiral oxidant such as a camphor-sulfonyl oxaziridine to generate the chiral sulfoxide intermediate.
- Oxidize the hydroxyl group of the chiral sulfoxide to an aldehyde using a sulfur trioxide-pyridine complex or similar oxidizing agent under controlled conditions.
- Execute the key cyclization step by treating the enantiomerically enriched aryl-3-propionaldehyde sulfoxide with a strong base like potassium tert-butoxide to close the seven-membered ring.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of the synthesis route described in CN1284953A offers tangible strategic benefits beyond mere technical elegance. The shift from biological or resolution-based methods to a fully chemical, stereocontrolled synthesis significantly de-risks the supply chain. Chemical processes are generally more predictable and easier to scale than fermentation-based ones, ensuring consistent delivery of high-purity pharmaceutical intermediates. The elimination of chiral chromatography steps removes a major cost driver and bottleneck, allowing for larger batch sizes and faster throughput times without the need for specialized preparative HPLC columns.
- Cost Reduction in Manufacturing: The process achieves cost optimization by removing the need for expensive chiral resolution steps and rare microbial cultures. By utilizing readily available chemical oxidants and bases, the raw material costs are stabilized and predictable. The high stereoselectivity means that less material is wasted as the unwanted enantiomer, effectively doubling the yield compared to a racemic synthesis followed by 50% loss during resolution. This efficiency translates into substantial cost savings per kilogram of active ingredient produced.
- Enhanced Supply Chain Reliability: Relying on commodity chemicals like potassium tert-butoxide, sodium hydride, and standard organic solvents ensures that the supply chain is not vulnerable to the shortages often associated with specialized biological reagents or chiral stationary phases. The robustness of the reaction conditions, which tolerate a range of temperatures and standard workup procedures, means that production can be easily transferred between different manufacturing sites if necessary, guaranteeing continuity of supply for critical cardiovascular medications.
- Scalability and Environmental Compliance: The synthetic route is designed for scalability, moving seamlessly from gram-scale laboratory experiments to multi-kilogram pilot runs and eventually to commercial tonnage production. The waste streams generated are primarily organic solvents and salts, which are well-understood and manageable within standard wastewater treatment protocols. Unlike biological processes that generate complex organic sludge, this chemical process allows for more straightforward solvent recovery and recycling, aligning with modern green chemistry principles and environmental regulations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of enantiomerically enriched tetrahydrobenzothiepine oxides. These answers are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy for R&D and quality assurance teams evaluating this technology for their pipelines.
Q: How is stereochemical control achieved at the C4 and C5 positions?
A: Stereocontrol is achieved through the chirality of the sulfur atom in the sulfoxide precursor. During the base-catalyzed cyclization, the existing chiral center at the sulfur atom directs the formation of the new chiral centers at the C4 and C5 positions of the seven-membered ring, ensuring high diastereoselectivity without the need for post-synthesis resolution.
Q: What are the advantages of this chemical synthesis over microbial methods?
A: Unlike microbial reduction methods which are limited to microgram scales and require complex culture maintenance, this chemical process utilizes standard organic reagents and conditions. This allows for straightforward scale-up from laboratory to commercial production (100 kgs to 100 MT) without the biological variability or purification challenges associated with fermentation broths.
Q: Can this process produce both (4R,5R) and (4S,5S) isomers?
A: Yes, the process is flexible. By selecting the appropriate enantiomer of the chiral oxidant (e.g., using either the (1R) or (1S) form of the camphor-sulfonyl oxaziridine) during the initial sulfide oxidation step, manufacturers can selectively produce either the (4R,5R) or (4S,5S) tetrahydrobenzothiepine oxide isomers as required for specific drug development programs.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tetrahydrobenzothiepine Oxide Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the development of life-saving cardiovascular therapies. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can move from clinical trials to market without supply interruptions. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of tetrahydrobenzothiepine oxide meets the exacting standards required for pharmaceutical applications, including precise control over enantiomeric excess and impurity profiles.
We invite you to collaborate with us to optimize your supply chain for IBAT inhibitors and related compounds. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and purity needs. Contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our advanced manufacturing capabilities can support your drug development goals efficiently and reliably.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →