Scalable Synthesis of Cilofexor Intermediates via Optimized Grignard Chemistry
Scalable Synthesis of Cilofexor Intermediates via Optimized Grignard Chemistry
The pharmaceutical landscape for treating non-alcoholic steatohepatitis (NASH) has been significantly advanced by the development of Farnesoid X Receptor (FXR) agonists, with Cilofexor (GS-9674) standing out as a prime candidate currently in clinical research. The efficient manufacturing of this complex molecule is critical for meeting future global demand. Patent CN115215853A discloses a groundbreaking preparation method for the compound of Formula I, offering a robust alternative to existing synthetic pathways. This technical insight report analyzes the novel methodology, which strategically bypasses the limitations of traditional noble metal catalysis and hazardous organolithium reagents. By leveraging optimized Grignard chemistry and mild nucleophilic substitution conditions, this process ensures high purity and operational safety, positioning it as a superior choice for commercial-scale production.
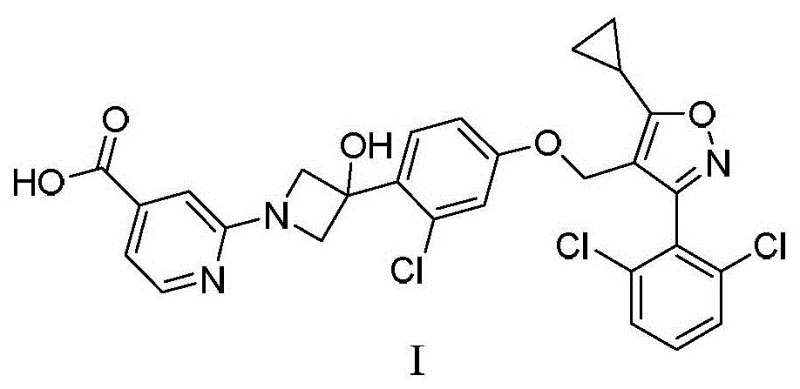
As a reliable pharmaceutical intermediate supplier, understanding the nuances of this synthetic route is essential for securing a stable supply chain. The disclosed method not only simplifies the reaction sequence but also enhances the overall economic viability of producing high-purity FXR agonists. The following sections provide a deep dive into the mechanistic advantages and commercial implications of this technology.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art synthesis routes, such as those reported in US20190142814A1 and US2017355693A1, present significant bottlenecks for large-scale manufacturing. These conventional pathways typically rely on palladium-catalyzed coupling reactions to construct the core biaryl or heteroaryl linkages. While effective on a small laboratory scale, the reliance on noble metal catalysts introduces substantial cost burdens due to the high price of palladium and the stringent requirements for removing trace metal residues to meet pharmaceutical standards. Furthermore, these routes often necessitate the use of n-butyllithium (n-BuLi) for halogen-lithium exchange steps.
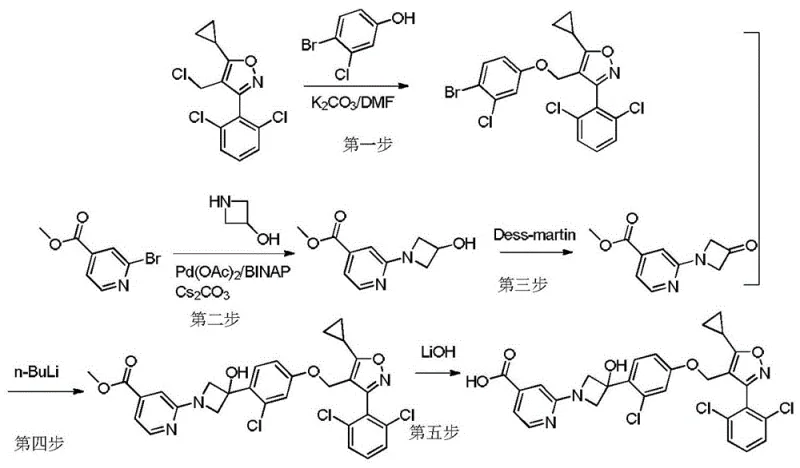
The use of n-BuLi imposes severe operational constraints, requiring cryogenic temperatures often as low as -78°C to maintain selectivity and prevent side reactions. Such harsh conditions are energy-intensive and difficult to control in large reactors, leading to safety risks and inconsistent batch quality. Additionally, patent data highlights that the fourth step in these traditional routes suffers from extremely low reaction yields, reported around 20 percent, which drastically reduces the overall throughput and increases waste generation. These factors collectively render conventional methods suboptimal for cost reduction in API manufacturing.
The Novel Approach
The methodology presented in CN115215853A represents a paradigm shift by eliminating both the noble metal catalyst and the hazardous n-BuLi reagent. Instead, the novel approach employs a Grignard reagent, specifically isopropyl magnesium bromide, to facilitate the key carbon-carbon bond formation. This substitution allows the reaction to proceed at much milder temperatures, ranging from -50°C to 15°C, which are far more manageable in an industrial setting. The avoidance of palladium not only lowers the direct material costs but also simplifies the downstream purification process by removing the need for specialized metal scavengers.
Moreover, the new route utilizes a straightforward nucleophilic substitution strategy for connecting the phenolic core to the isoxazole side chain. By optimizing the base and solvent system, the process achieves high conversion rates without the need for complex catalytic cycles. This streamlined approach results in a shorter reaction sequence with fewer unit operations, directly translating to improved process efficiency. The ability to operate under less extreme conditions while maintaining high yields makes this novel approach highly attractive for the commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Isopropyl Grignard Addition and Substitution
The core innovation of this synthesis lies in the strategic application of Grignard chemistry to construct the azetidine scaffold. In Step D of the disclosed process, the aryl halide intermediate (Compound 3) undergoes a halogen-metal exchange using isopropyl magnesium bromide. Unlike the highly reactive and pyrophoric n-BuLi, the Grignard reagent offers a more controlled reactivity profile. The resulting organomagnesium species then attacks the carbonyl group of the protected azetidinone (Compound 4), forming the tertiary alcohol center with high stereochemical integrity. This step is critical as it establishes the chiral center necessary for biological activity.
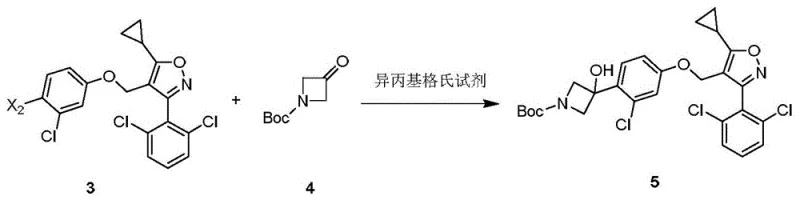
The patent data indicates that the choice of halogen on the aromatic ring significantly influences the reaction efficiency. When the leaving group X2 is iodine, the conversion rate of raw materials reaches up to 96 percent at -20°C, whereas bromine analogues show markedly lower conversion under similar conditions. This mechanistic insight allows process chemists to select the optimal starting material to maximize yield. Furthermore, the reaction tolerates a range of temperatures from -50°C to 15°C, providing flexibility in thermal management during scale-up. The use of tetrahydrofuran (THF) as a solvent stabilizes the Grignard reagent, ensuring consistent performance throughout the addition phase.
Following the Grignard addition, the synthesis proceeds through a deprotection and substitution sequence. The removal of the Boc protecting group using p-toluenesulfonic acid reveals the secondary amine, which then acts as a nucleophile in Step A. Here, the amine attacks the activated pyridine derivative (Compound 7) in the presence of a mild base like potassium carbonate. This SNAr-type substitution is highly efficient, with HPLC analysis showing target product content of up to 86 percent in the reaction mixture with negligible byproduct formation. The mechanistic simplicity of this step ensures that the final coupling is robust and reproducible, minimizing the risk of batch failures.
How to Synthesize Cilofexor Efficiently
The synthesis of Cilofexor intermediates via this novel route involves a logical sequence of five key transformations that prioritize safety and yield. The process begins with the etherification of the isoxazole core, followed by the critical Grignard-mediated addition to the azetidinone ring. Subsequent deprotection exposes the amine functionality required for the final coupling with the pyridine ester. The sequence concludes with a mild hydrolysis step to reveal the free carboxylic acid. Each step has been optimized to avoid extreme conditions, making the entire pathway suitable for transfer from pilot plant to full commercial production. For detailed operational parameters, stoichiometry, and workup procedures, please refer to the standardized guide below.
- Perform etherification of compound 1 with compound 2a using potassium carbonate in DMF at 80°C to obtain compound 3a.
- React compound 3a with isopropyl magnesium bromide followed by compound 4 at -20°C to -30°C to form the azetidine intermediate compound 5.
- Remove the Boc protecting group from compound 5 using p-toluenesulfonic acid in dichloromethane to yield compound 6.
- Couple compound 6 with compound 7a using potassium carbonate in DMF at 70°C to generate the ester intermediate compound 8a.
- Hydrolyze compound 8a using lithium hydroxide monohydrate in a THF/methanol/water mixture at room temperature to afford Cilofexor (Formula I).
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthesis route offers tangible strategic benefits beyond mere technical feasibility. The primary advantage lies in the drastic simplification of the supply chain for raw materials. By eliminating the need for palladium catalysts and n-BuLi, manufacturers can source reagents that are commodity chemicals rather than specialized, high-cost items. This shift reduces exposure to price volatility in the precious metals market and mitigates the risks associated with the storage and handling of pyrophoric substances. Consequently, the overall cost of goods sold (COGS) is significantly reduced, enhancing the margin potential for the final API.
- Cost Reduction in Manufacturing: The elimination of noble metal catalysts removes the necessity for expensive metal scavenging resins and extensive filtration steps, which are traditionally required to meet strict residual metal limits in pharmaceuticals. Furthermore, the replacement of n-BuLi with isopropyl magnesium bromide reduces the energy consumption associated with maintaining cryogenic temperatures. The higher yields observed in the Grignard step compared to prior art mean that less raw material is wasted per kilogram of product, leading to substantial cost savings in material usage. These cumulative efficiencies result in a more economically competitive manufacturing process.
- Enhanced Supply Chain Reliability: The reagents utilized in this pathway, such as potassium carbonate, THF, and isopropyl magnesium bromide, are widely available from multiple global suppliers, ensuring a resilient supply chain. Unlike specialized catalysts that may have long lead times or single-source dependencies, these commodity chemicals can be procured with short notice. The robustness of the reaction conditions also means that production schedules are less likely to be disrupted by minor fluctuations in environmental controls or reagent quality. This reliability is crucial for maintaining continuous supply to downstream API manufacturers.
- Scalability and Environmental Compliance: The process operates under milder thermal conditions and generates fewer hazardous byproducts, simplifying waste treatment and disposal. The absence of heavy metals reduces the environmental footprint of the manufacturing site and eases regulatory compliance burdens. The high conversion rates and purity profiles demonstrated in the patent examples indicate that the process is inherently scalable, allowing for seamless transition from kilogram to ton-scale production without significant re-engineering. This scalability ensures that supply can be rapidly ramped up to meet clinical or commercial demand surges.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis of Cilofexor intermediates as described in patent CN115215853A. These answers are derived directly from the experimental data and process descriptions provided in the intellectual property documentation. They serve to clarify the operational advantages and quality metrics associated with this specific manufacturing route. Understanding these details is vital for stakeholders evaluating the feasibility of adopting this technology for their supply chains.
Q: Why is the Grignard reagent preferred over n-BuLi in this synthesis?
A: The use of isopropyl magnesium bromide avoids the harsh cryogenic conditions (-78°C) and safety hazards associated with n-BuLi. Patent data indicates significantly higher conversion rates (up to 96%) compared to the 20% yield often seen with n-BuLi routes, ensuring better scalability.
Q: What is the achieved purity of the final Cilofexor product?
A: According to Example 5 in the patent data, the final hydrolysis step yields Cilofexor with a purity of 99.8% and an isolated yield of 93.42%, demonstrating the high efficiency of the purification protocol.
Q: Does this process require expensive noble metal catalysts?
A: No. Unlike prior art methods that rely on palladium catalysis for coupling, this novel route utilizes standard nucleophilic substitution with inorganic bases like potassium carbonate, drastically reducing raw material costs and eliminating heavy metal contamination risks.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Cilofexor Supplier
The technological advancements detailed in this report underscore the potential for efficient, high-quality production of FXR agonist intermediates. At NINGBO INNO PHARMCHEM, we possess the technical expertise and infrastructure to translate these innovative synthetic routes into commercial reality. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met with precision and speed. We adhere to stringent purity specifications and utilize rigorous QC labs to guarantee that every batch meets the highest international standards for pharmaceutical intermediates.
We invite you to collaborate with us to leverage this optimized synthesis for your Cilofexor supply chain. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Please contact us to request specific COA data and route feasibility assessments, and let us demonstrate how our manufacturing capabilities can drive value and security for your organization.