Advanced Manufacturing of N-Substituted Thiomorpholine Derivatives for Next-Generation Diabetes Therapeutics
Advanced Manufacturing of N-Substituted Thiomorpholine Derivatives for Next-Generation Diabetes Therapeutics
The pharmaceutical landscape for Type 2 diabetes management has been significantly transformed by the advent of Dipeptidyl Peptidase-IV (DPP-IV) inhibitors, which function by prolonging the activity of incretin hormones such as GLP-1 and GIP. Patent CN101279955B discloses a novel class of N-substituted thiomorpholine compounds that exhibit potent inhibitory activity against DPP-IV, offering a robust structural scaffold for drug development. These compounds are characterized by a chiral thiomorpholine ring substituted with a cyano group at the 3-position and an amino acid moiety at the nitrogen atom, a configuration that is critical for high-affinity binding to the enzyme active site. The structural versatility of this scaffold allows for extensive modification of the amino acid side chain, enabling the fine-tuning of pharmacokinetic properties and metabolic stability. As a reliable pharmaceutical intermediates supplier, understanding the nuances of this specific chemical architecture is essential for developing cost-effective and high-purity supply chains. The following analysis delves into the synthetic methodology and commercial viability of these advanced therapeutic agents.
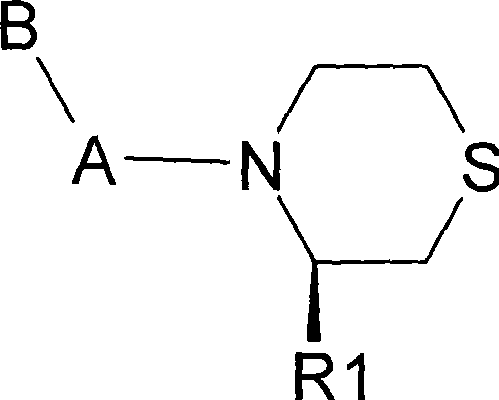
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes to thiomorpholine derivatives often suffer from significant drawbacks regarding stereochemical control and process efficiency. Many conventional methods rely on the alkylation of achiral thiomorpholine precursors, which inevitably leads to the formation of racemic mixtures requiring expensive and yield-loss-inducing chiral resolution steps. Furthermore, the introduction of the critical nitrile functionality at the 3-position frequently involves harsh dehydration conditions or toxic cyanide sources, posing severe safety hazards and environmental compliance challenges in a commercial setting. The lack of stereospecificity in these older routes not only complicates the purification process but also raises regulatory concerns regarding the presence of potentially inactive or toxic enantiomers in the final drug substance. Additionally, multi-step sequences involving protecting group manipulations often result in cumulative yield losses, driving up the cost of goods sold (COGS) and limiting the economic feasibility of large-scale production.
The Novel Approach
The methodology outlined in the patent data presents a paradigm shift by utilizing L-cysteine, a naturally occurring chiral amino acid, as the starting material to construct the thiomorpholine ring with inherent stereocontrol. This biomimetic approach ensures that the chirality at the 3-position is established early in the synthesis and maintained throughout the subsequent transformations, effectively eliminating the need for downstream chiral separation. The process employs a stereospecific intramolecular nucleophilic substitution to close the ring, a reaction that proceeds with high fidelity under mild aqueous conditions. Moreover, the conversion of the amide intermediate to the nitrile group is achieved using trifluoroacetic anhydride (TFAA), a reagent that offers a safer and more controllable alternative to traditional phosphorus oxychloride or thionyl chloride dehydration methods. This novel route not only enhances the overall optical purity of the product but also streamlines the manufacturing process, making it highly attractive for cost reduction in pharmaceutical intermediates manufacturing.
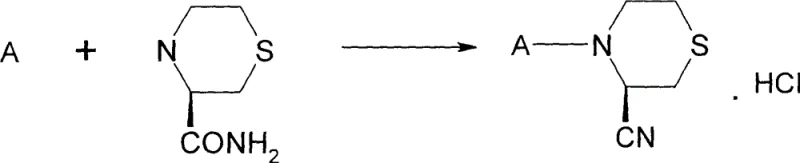
Mechanistic Insights into Stereospecific Cyclization and Dehydration
The core of this synthetic strategy lies in the construction of the chiral thiomorpholine scaffold from L-cysteine derivatives through a carefully orchestrated sequence of functional group interconversions. The process begins with the reaction of L-cysteine with ethylene oxide to form 2-hydroxyethylcysteine, followed by chlorination to generate the corresponding chloroethyl derivative. The pivotal step is the base-mediated intramolecular cyclization of the methyl ester of 2-chloroethylcysteine, where the amino group acts as a nucleophile to displace the chloride ion, forming the six-membered thiomorpholine ring. This cyclization is stereospecific, preserving the configuration of the alpha-carbon from the original cysteine, which is crucial for the biological activity of the final DPP-IV inhibitor. The reaction is typically conducted in water with a mild base such as sodium bicarbonate, which minimizes side reactions and simplifies the workup procedure, thereby enhancing the robustness of the process for industrial application.
Following the formation of the thiomorpholine ring, the ester group is converted to a primary amide via ammonolysis, setting the stage for the installation of the warhead nitrile group. The dehydration of this amide to the nitrile is facilitated by activation with trifluoroacetic anhydride, which generates a reactive imidate intermediate that eliminates trifluoroacetic acid to yield the cyano functionality. This mechanism is particularly advantageous because it proceeds under relatively mild conditions compared to traditional dehydrating agents, reducing the risk of epimerization at the adjacent chiral center. The final step involves peptide coupling with a BOC-protected amino acid using standard carbodiimide chemistry (EDCI/HOBt), followed by acidic deprotection to reveal the free amine. This modular approach allows for the facile incorporation of various amino acid side chains, providing a versatile platform for structure-activity relationship (SAR) studies and optimization of the drug candidate.
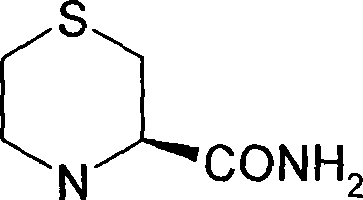
How to Synthesize (R)-3-Cyano-4-(aminoacyl)thiomorpholine Efficiently
The synthesis of these high-value intermediates requires precise control over reaction parameters to ensure consistent quality and yield. The process initiates with the preparation of the chiral thiomorpholine core, where strict temperature control during the cyclization step is vital to prevent the formation of byproducts. Subsequent amidation and dehydration steps must be monitored closely to ensure complete conversion while avoiding over-reaction or degradation of the sensitive heterocyclic ring. The final coupling reaction demands anhydrous conditions to maximize the efficiency of the peptide bond formation. For a detailed breakdown of the specific operational parameters, reagent stoichiometry, and purification techniques required to execute this synthesis successfully, please refer to the standardized protocol below.
- Prepare the chiral thiomorpholine core by reacting L-cysteine derivatives with ethylene oxide followed by chlorination and stereospecific ring closure in aqueous media.
- Convert the resulting ester intermediate into the primary amide through ammonolysis in methanol to establish the necessary functionality for dehydration.
- Perform peptide coupling with BOC-protected amino acids using EDCI/HOBt, followed by dehydration with trifluoroacetic anhydride and final deprotection to yield the target inhibitor.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the synthetic route described in patent CN101279955B offers substantial strategic advantages over legacy manufacturing methods. The reliance on L-cysteine as a starting material leverages a widely available and cost-effective commodity chemical, ensuring a stable and resilient supply chain that is less susceptible to market volatility. The elimination of chiral resolution steps significantly reduces the number of unit operations and solvent consumption, leading to a drastically simplified process flow that translates directly into lower manufacturing costs and reduced waste generation. Furthermore, the use of aqueous media for the critical ring-closing step aligns with green chemistry principles, minimizing the environmental footprint and facilitating easier regulatory approval for commercial scale-up of complex pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The process achieves significant cost optimization by utilizing inexpensive, naturally sourced chiral pool materials rather than costly chiral catalysts or resolving agents. The high atom economy of the cyclization reaction and the avoidance of hazardous reagents like cyanide salts further contribute to reduced raw material and waste disposal expenses. By streamlining the synthesis into fewer steps with high-yielding transformations, the overall production efficiency is markedly improved, allowing for competitive pricing strategies in the global market.
- Enhanced Supply Chain Reliability: Sourcing L-cysteine and standard peptide coupling reagents ensures a diversified and robust supply base, mitigating the risks associated with single-source dependencies on exotic reagents. The robustness of the aqueous cyclization step makes the process less sensitive to minor variations in raw material quality, ensuring consistent batch-to-batch reproducibility. This reliability is critical for maintaining continuous production schedules and meeting the stringent delivery timelines required by multinational pharmaceutical clients.
- Scalability and Environmental Compliance: The synthetic route is inherently scalable, as demonstrated by the successful execution of key steps in water and common organic solvents like THF and ethyl acetate. The absence of heavy metal catalysts simplifies the purification process and ensures that the final product meets rigorous impurity specifications without the need for complex metal scavenging technologies. This environmental compatibility facilitates smoother regulatory filings and supports sustainable manufacturing practices, which are increasingly becoming a prerequisite for supplier qualification in the modern pharmaceutical industry.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these thiomorpholine derivatives. The answers are derived directly from the patented technology and reflect the current best practices in the field of DPP-IV inhibitor synthesis. Understanding these details is crucial for stakeholders evaluating the feasibility of integrating this chemistry into their development pipelines.
Q: What is the key advantage of the cysteine-based route for thiomorpholine synthesis?
A: The cysteine-based route offers inherent chirality from the natural amino acid source, ensuring high stereospecificity during the ring-closing step without the need for complex chiral resolution processes.
Q: How is the nitrile group introduced in the final structure?
A: The nitrile group is generated via a mild dehydration of the primary amide intermediate using trifluoroacetic anhydride (TFAA), which avoids harsh conditions that could compromise the sensitive thiomorpholine ring.
Q: Is this synthesis suitable for large-scale commercial production?
A: Yes, the process utilizes commodity chemicals like L-cysteine and standard peptide coupling reagents, and the ring closure occurs in water, making it highly scalable and environmentally compliant for industrial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable N-Substituted Thiomorpholine Supplier
At NINGBO INNO PHARMCHEM, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from laboratory discovery to market launch is seamless and efficient. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of N-substituted thiomorpholine intermediates meets the highest international standards. We understand the critical nature of chiral purity in DPP-IV inhibitors and employ advanced analytical techniques to monitor and control stereochemistry throughout the manufacturing process. Our commitment to quality and consistency makes us the preferred partner for pharmaceutical companies seeking to secure their supply of critical diabetes therapeutics.
We invite you to engage with our technical procurement team to discuss how we can tailor our manufacturing capabilities to your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain valuable insights into how our optimized synthetic route can reduce your overall development costs. We encourage potential partners to contact us for specific COA data and route feasibility assessments to verify our capability to deliver high-quality intermediates that accelerate your drug development timeline. Let us collaborate to bring next-generation diabetes treatments to patients faster and more efficiently.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →