Scalable Synthesis of 4-Demethoxy-4-Aminoanthracycline Intermediates for Advanced Oncology Applications
Scalable Synthesis of 4-Demethoxy-4-Aminoanthracycline Intermediates for Advanced Oncology Applications
The development of next-generation antineoplastic agents requires precise chemical engineering to overcome limitations such as multidrug resistance and cardiotoxicity associated with classical anthracyclines. Patent CN1022038C discloses a sophisticated preparation method for 4-demethoxy-4-aminoanthracycline compounds, a class of molecules structurally related to daunorubicin but modified to enhance therapeutic indices. This technology leverages a novel nordaunosaminone derivative pathway, initiating from natural carminomycinone, to construct the critical 4-amino functionality with high regioselectivity. For R&D directors and procurement specialists in the oncology sector, understanding this synthetic route is vital for securing reliable sources of high-purity intermediates that serve as the backbone for potent anticancer drugs. The process elegantly combines selective protection strategies with efficient nucleophilic substitutions, offering a robust alternative to total synthesis.
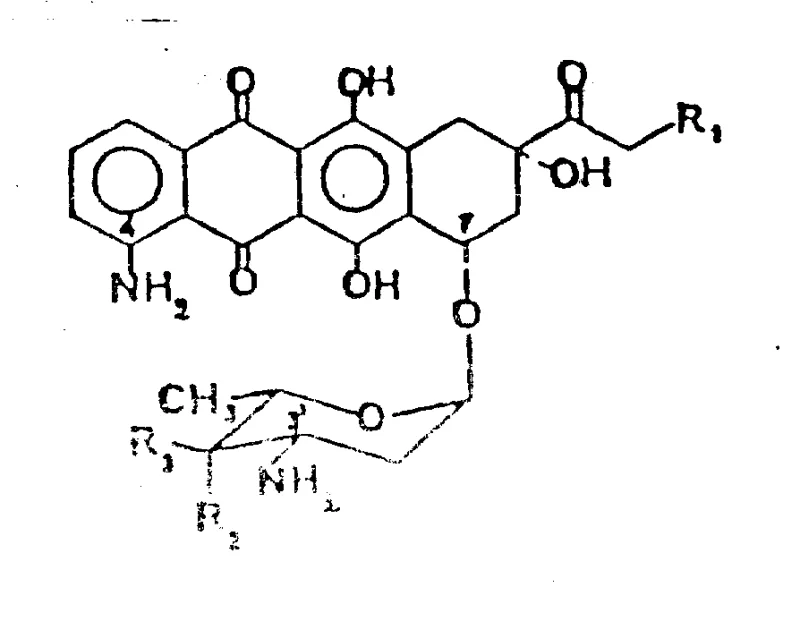
In the competitive landscape of pharmaceutical intermediates, the ability to consistently produce complex glycosides with defined stereochemistry is a significant differentiator. The disclosed method targets compounds of Formula I, where the sugar moiety is coupled to an aglycone core that has been meticulously functionalized at the C-4 and C-7 positions. By starting from naturally derived precursors and employing specific activation steps, the process minimizes the generation of difficult-to-remove isomers. This level of control is paramount for meeting the stringent purity specifications required by global regulatory bodies for clinical trial materials and commercial API production. Furthermore, the versatility of the route allows for the preparation of various acid addition salts, such as hydrochlorides, facilitating formulation development.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional approaches to synthesizing 4-aminoanthracyclines often struggle with the inherent reactivity of the anthraquinone nucleus. Direct amination of demethoxylated precursors can lead to over-reaction, polymerization, or degradation of the sensitive quinone system due to the harsh conditions typically required to displace a methoxy or hydroxyl group. Additionally, achieving selectivity between the multiple hydroxyl groups present on the aglycone (at positions 6, 11, and 9) without extensive protection-deprotection sequences is chemically challenging. Conventional routes may also suffer from poor stereocontrol during the glycosylation step, resulting in mixtures of alpha and beta anomers that are costly and time-consuming to separate. These inefficiencies translate directly into higher manufacturing costs and longer lead times, creating bottlenecks in the supply chain for critical cancer therapies.
The Novel Approach
The methodology outlined in the patent introduces a transformative strategy by utilizing a sulfonate activation mechanism. Instead of attempting direct displacement, the process converts the C-4 hydroxyl group into a 4-O-(4-fluorobenzenesulfonyl) derivative (Formula VII). This transformation dramatically increases the leaving group ability at the C-4 position, enabling nucleophilic attack by amines under much milder conditions. As illustrated in the comprehensive reaction scheme below, this allows for the clean introduction of the amino group via a benzylamine intermediate, which is subsequently removed by catalytic hydrogenation. This stepwise approach preserves the integrity of the anthraquinone chromophore and ensures that the C-6 and C-11 phenolic hydroxyls remain unaffected, demonstrating exceptional chemoselectivity.
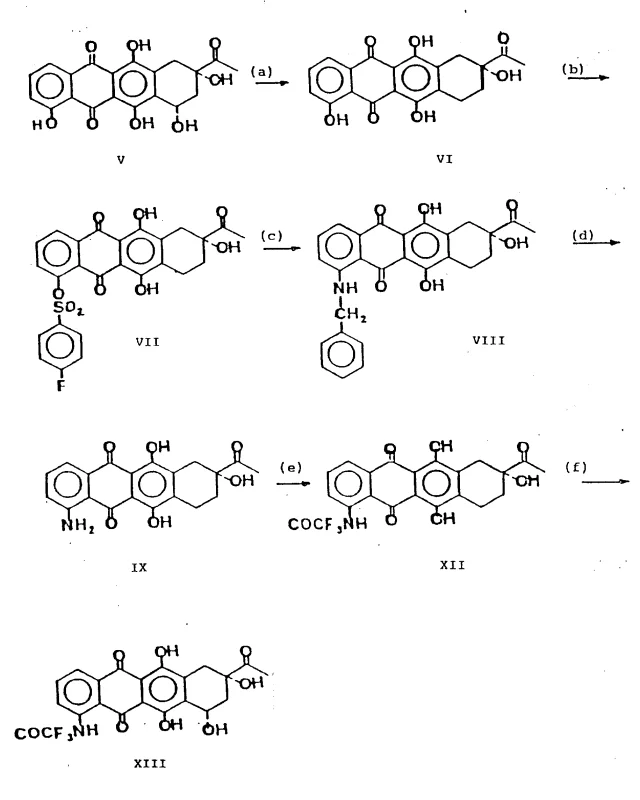
Furthermore, the novel approach integrates a highly effective protection strategy for the amino group using trifluoroacetyl moieties. This temporary masking prevents unwanted side reactions during the subsequent glycosylation coupling with the sugar derivative. The use of silver trifluoromethanesulfonate as a promoter in the glycosylation step ensures the formation of the desired alpha-glycosidic linkage with high fidelity. By decoupling the amination and glycosylation events through stable intermediates, the process achieves a level of modularity that is highly advantageous for scale-up. This contrasts sharply with one-pot methods that often sacrifice yield for speed, providing a more reliable pathway for generating the complex molecular architecture required for biological activity.
Mechanistic Insights into Sulfonate-Mediated Nucleophilic Substitution
The core chemical innovation in this synthesis lies in the activation of the C-4 position via sulfonation. In the anthracycline structure, the C-4 hydroxyl is allylic to the quinone system, which can influence its reactivity. However, converting this hydroxyl into a 4-fluorobenzenesulfonate ester creates a superior leaving group compared to halides or tosylates in this specific steric environment. The electron-withdrawing nature of the fluorine atom on the sulfonyl ring further enhances the electrophilicity of the sulfur center, facilitating the departure of the sulfonate group when attacked by the nucleophilic nitrogen of benzylamine. This reaction likely proceeds through an SN2-like mechanism where the amine attacks the C-4 carbon from the less hindered face, dictated by the conformation of the saturated ring D. The result is the inversion or retention of configuration depending on the specific transition state, but critically, it occurs without disrupting the adjacent carbonyl at C-13 or the phenolic systems.
Impurity control is rigorously managed through the selection of protecting groups. The use of the trifluoroacetyl group for the amine (forming Formula XII and XIII) is particularly strategic. Trifluoroacetamides are stable enough to withstand the acidic conditions of glycosylation yet can be removed selectively under mild alkaline hydrolysis. This orthogonality is essential for preventing the formation of N-glycosylated byproducts, which are common impurities in anthracycline synthesis. Additionally, the initial hydrogenolysis step to remove the 7-alpha-hydroxy group (converting V to VI) simplifies the molecule early in the sequence. By eliminating this stereocenter before the critical C-4 functionalization, the process reduces the complexity of the diastereomeric mixture, ensuring that the final product possesses the correct 7S, 9S configuration required for DNA intercalation and topoisomerase II inhibition.
How to Synthesize 4-Demethoxy-4-Aminoanthracycline Efficiently
The synthesis of these potent antineoplastic intermediates requires strict adherence to anhydrous conditions and precise temperature control, particularly during the activation and coupling phases. The process begins with the modification of the natural product carminomycinone, leveraging its existing chiral pool to establish the core stereochemistry. Operators must ensure that the sulfonation step is quenched effectively to prevent over-sulfonation of the phenolic hydroxyls, although the patent notes surprising selectivity for the C-4 position under the described conditions. Following the amination and deprotection sequence, the resulting 4-amino-7-deoxyaglycone is ready for the final glycosylation. Detailed standard operating procedures for each reaction stage, including workup and purification protocols, are essential for reproducibility.
- Perform hydrogenolysis on Carminomycinone (Formula V) to remove the 7-alpha-hydroxy group, yielding 4-demethoxy-7-deoxydaunomycinone (Formula VI).
- React Formula VI with 4-fluorobenzenesulfonyl chloride to selectively form the C-4-O-sulfonyl derivative (Formula VII), activating the position for nucleophilic attack.
- Execute nucleophilic substitution using benzylamine to introduce the amino group, followed by catalytic hydrogenation to remove the benzyl protecting group, yielding the 4-amino derivative (Formula IX).
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers tangible benefits beyond mere chemical elegance. The reliance on carminomycinone as a starting material taps into a renewable biological source, reducing dependency on petrochemical feedstocks and mitigating risks associated with volatile raw material markets. The stepwise nature of the synthesis allows for quality checkpoints at each intermediate stage (VI, VII, VIII, IX), enabling early detection of deviations and preventing the loss of value-added materials in later steps. This modular approach significantly enhances supply chain resilience, as intermediates can potentially be stocked or sourced from different vendors if necessary, ensuring continuity of supply for the final API.
- Cost Reduction in Manufacturing: The elimination of harsh reagents and the use of catalytic hydrogenation for deprotection steps contribute to a safer and more cost-effective process profile. By avoiding expensive transition metal catalysts often required for direct C-N bond formation in similar contexts, the method reduces both material costs and the burden of heavy metal clearance testing. The high selectivity of the sulfonate displacement minimizes the formation of byproducts, leading to improved overall yields and reduced solvent consumption for purification. These efficiencies aggregate to lower the cost of goods sold (COGS), making the final therapeutic more accessible while maintaining healthy margins for manufacturers.
- Enhanced Supply Chain Reliability: The reagents utilized, such as 4-fluorobenzenesulfonyl chloride and benzylamine, are commodity chemicals available from multiple global suppliers, reducing single-source dependency risks. The robustness of the trifluoroacetyl protection strategy ensures that the intermediates have sufficient shelf-life for transport and storage, facilitating international logistics. Furthermore, the process avoids cryogenic conditions or ultra-high pressure equipment, allowing production in standard multipurpose chemical plants. This flexibility enables rapid scale-up from pilot to commercial volumes, ensuring that demand surges for oncology treatments can be met without lengthy lead times for new equipment installation.
- Scalability and Environmental Compliance: The waste streams generated by this process are manageable and amenable to standard treatment protocols. The use of organic solvents like methylene chloride and tetrahydrofuran is well-established in the industry, with mature recovery and recycling infrastructure available. The avoidance of stoichiometric heavy metal oxidants or toxic cyanide sources aligns with modern green chemistry principles and increasingly stringent environmental regulations. This compliance reduces the regulatory burden on manufacturing sites and minimizes the risk of production shutdowns due to environmental non-compliance, securing long-term operational stability for the supply chain.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of 4-demethoxy-4-aminoanthracycline intermediates. These insights are derived directly from the patented technology and are intended to clarify the feasibility and advantages of this specific synthetic pathway for potential partners and licensees. Understanding these details is crucial for making informed decisions about process adoption and integration into existing manufacturing portfolios.
Q: What is the primary advantage of using the sulfonate intermediate (Formula VII) in this synthesis?
A: The formation of the 4-O-(4-fluorobenzenesulfonyl) derivative significantly enhances the leaving group ability at the C-4 position. This allows for highly regioselective nucleophilic substitution by amines under mild conditions, avoiding the harsh reagents typically required for direct amination of anthraquinones, thereby preserving the sensitive quinone moiety.
Q: How does this method address the issue of drug resistance in anthracycline therapy?
A: The resulting 4-demethoxy-4-aminoanthracycline compounds lack the 4-methoxy group found in traditional agents like daunorubicin. Structural modifications at the C-4 position are known to reduce recognition by P-glycoprotein efflux pumps, potentially overcoming multidrug resistance (MDR) in tumor cells while maintaining potent cytotoxicity.
Q: Is the glycosylation step compatible with large-scale manufacturing?
A: Yes, the described glycosylation utilizes silver trifluoromethanesulfonate as a promoter in anhydrous methylene chloride. While silver salts require recovery protocols, the reaction proceeds at low temperatures (5-10°C) with high stereocontrol, minimizing the formation of beta-anomer impurities and simplifying downstream purification processes essential for GMP production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 4-Demethoxy-4-Aminoanthracycline Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the development of life-saving oncology drugs. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated chemistry described in patent CN1022038C can be translated into industrial reality. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify the identity and potency of every batch. Our commitment to quality assurance means that you receive materials that are ready for the next stage of synthesis, whether it be final API manufacturing or preclinical formulation studies.
We invite you to collaborate with us to optimize your supply chain for anthracycline-based therapeutics. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. Contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our advanced manufacturing capabilities can support your mission to bring innovative cancer treatments to patients worldwide.