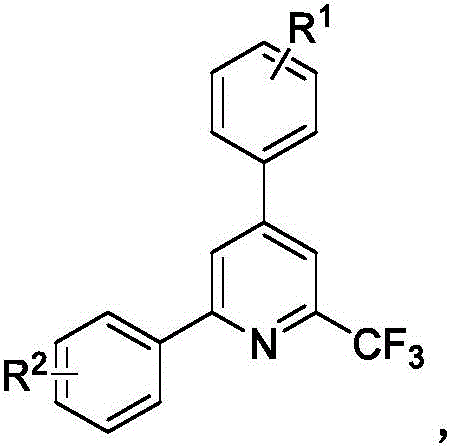
Scalable Synthesis of 2,4-Diaryl-6-Trifluoromethylpyridine Derivatives for Commercial API Manufacturing
Scalable Synthesis of 2,4-Diaryl-6-Trifluoromethylpyridine Derivatives for Commercial API Manufacturing
The pharmaceutical and fine chemical industries are constantly seeking robust methodologies for constructing fluorinated heterocyclic scaffolds, which are critical motifs in modern drug design. Patent CN112174877B discloses a groundbreaking preparation method for 2,4-diaryl-6-trifluoromethylpyridine derivatives, addressing the longstanding challenges associated with introducing trifluoromethyl groups into pyridine cores. This technology leverages a novel copper-catalyzed cyclization strategy that bypasses the limitations of traditional cross-coupling reactions. By utilizing readily available alkenyl azide compounds and trifluoromethyl alkynone derivatives, this process achieves high yields under remarkably mild conditions. The significance of this innovation lies in its ability to access diverse substitution patterns that were previously difficult or impossible to synthesize efficiently. For R&D directors and procurement specialists, this represents a pivotal shift towards more sustainable and cost-effective manufacturing pathways for high-value pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of trifluoromethyl-substituted pyridine rings has relied heavily on transition metal-mediated cross-coupling reactions involving pre-functionalized pyridine precursors. These conventional pathways are fraught with significant inefficiencies that hinder commercial viability. Firstly, they often suffer from poor regioselectivity, leading to complex mixtures of by-products that require extensive and costly purification efforts to isolate the desired isomer. Secondly, the reliance on precious transition metal catalysts, such as palladium or platinum complexes, imposes a substantial financial burden on the manufacturing process, particularly when scaling to metric ton quantities. Furthermore, these methods typically necessitate harsh reaction conditions, including elevated temperatures and stringent anhydrous environments, which increase energy consumption and equipment requirements. The need for pre-functionalization of the pyridine skeleton also results in poor atom economy, generating excessive chemical waste and complicating the supply chain with additional synthetic steps.
The Novel Approach
In stark contrast, the methodology outlined in CN112174877B introduces a streamlined direct cyclization approach that fundamentally reshapes the synthetic landscape for these derivatives. This novel route utilizes a copper-catalyzed reaction between alkenyl azides and trifluoromethyl alkynones, effectively assembling the pyridine ring in a single pot. The process operates at ambient to slightly elevated temperatures ranging from 25°C to 35°C, drastically reducing energy inputs compared to traditional high-temperature protocols. By employing earth-abundant copper salts instead of precious metals, the method significantly lowers the raw material costs and simplifies the removal of metal residues from the final product. The reaction demonstrates exceptional functional group tolerance, accommodating a wide array of substituents including halogens, alkyl groups, and alkoxy groups without compromising yield or selectivity. This versatility allows for the rapid generation of diverse chemical libraries, accelerating the drug discovery process while maintaining a green chemistry profile through simplified post-processing.
Mechanistic Insights into Copper-Catalyzed Cyclization
The core of this technological breakthrough lies in the intricate copper-catalyzed mechanism that facilitates the formation of the pyridine ring from acyclic precursors. The reaction initiates with the activation of the alkenyl azide by the phosphine additive, likely forming a reactive iminophosphorane intermediate in situ. This species then engages with the electron-deficient trifluoromethyl alkynone in a cycloaddition-like process mediated by the copper catalyst. The copper center plays a dual role, coordinating to the alkyne moiety to enhance its electrophilicity while simultaneously stabilizing the developing negative charge during the ring-closing event. This cooperative catalysis ensures high regioselectivity, directing the cyclization to exclusively form the 2,4-diaryl-6-trifluoromethyl substitution pattern. The mild basic conditions provided by amines such as pentamethyldiethylenetriamine facilitate the final aromatization step, driving the equilibrium towards the stable pyridine product. Understanding this mechanism is crucial for process chemists aiming to optimize reaction parameters for specific substrate classes.
From an impurity control perspective, the specificity of this catalytic cycle offers distinct advantages over radical-based or high-energy thermal methods. The controlled nature of the copper coordination sphere minimizes side reactions such as polymerization of the alkyne or decomposition of the azide functionality. The use of mild bases prevents the hydrolysis of sensitive functional groups that might be present on the aryl rings, ensuring a cleaner crude reaction profile. Consequently, the burden on downstream purification is significantly reduced, as the primary impurities are often unreacted starting materials rather than structurally similar by-products. This high level of chemoselectivity is paramount for pharmaceutical applications where strict limits on genotoxic impurities and heavy metals are enforced. The ability to achieve high purity directly from the reaction mixture translates to higher overall process efficiency and reduced solvent consumption during the isolation phase.
How to Synthesize 2,4-Diaryl-6-Trifluoromethylpyridine Efficiently
The practical implementation of this synthesis involves a straightforward two-stage addition protocol that is easily adaptable to standard reactor setups. Initially, the alkenyl azide is activated in a non-polar solvent like toluene with a phosphine additive, establishing the reactive intermediate necessary for cyclization. Subsequently, the introduction of the polar co-solvent DMSO along with the copper catalyst and base creates the optimal environment for the coupling with the trifluoromethyl alkynone. The careful control of addition rates and temperature during the second stage is critical to managing the exotherm and ensuring uniform mixing. Detailed standardized synthesis steps see the guide below.
- Mix alkenyl azide compound with additive and first organic solvent, reacting at 25-35°C for 0.4-0.6 hours.
- Add base, catalyst, and second organic solvent, then slowly dropwise add trifluoromethyl alkynone derivative in an ice bath before reacting at 25-35°C for 12-24 hours.
- Extract the reaction mixture with water and ethyl acetate, concentrate the organic phase, and purify via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers compelling economic and logistical benefits that extend beyond simple yield metrics. The elimination of expensive precious metal catalysts directly impacts the bill of materials, resulting in substantial cost savings that improve the margin profile of the final active pharmaceutical ingredient. Moreover, the use of commodity chemicals such as copper bromide and common organic solvents ensures a stable and resilient supply chain, mitigating the risks associated with the volatility of rare metal markets. The mild reaction conditions reduce the dependency on specialized high-pressure or high-temperature equipment, allowing for production in existing multipurpose facilities without significant capital expenditure. This flexibility enhances manufacturing agility, enabling faster response times to market demands and shorter lead times for custom synthesis projects.
- Cost Reduction in Manufacturing: The replacement of palladium or platinum catalysts with inexpensive copper salts represents a direct reduction in catalyst costs, which is a major driver of overall production expenses. Additionally, the high atom economy of the cyclization reaction minimizes waste disposal costs and maximizes the utilization of raw materials. The simplified workup procedure, involving standard aqueous extraction and chromatography, reduces labor hours and solvent usage compared to complex multi-step purifications required by older methods. These cumulative efficiencies translate into a lower cost of goods sold, providing a competitive pricing advantage in the global marketplace for pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: The starting materials, specifically the alkenyl azides and trifluoromethyl alkynones, are derived from widely available bulk chemicals, ensuring consistent availability and reducing the risk of supply disruptions. The robustness of the reaction conditions means that the process is less sensitive to minor variations in raw material quality or environmental factors, leading to more predictable batch-to-batch consistency. This reliability is critical for maintaining continuous production schedules and meeting strict delivery commitments to downstream clients. Furthermore, the scalability of the process from gram to kilogram scales has been demonstrated, confirming its viability for commercial manufacturing without the need for extensive re-optimization.
- Scalability and Environmental Compliance: The green chemistry attributes of this method align perfectly with increasingly stringent environmental regulations and corporate sustainability goals. The reduction in hazardous waste generation and energy consumption lowers the environmental footprint of the manufacturing process. The absence of toxic heavy metals simplifies the regulatory approval process for new drug applications, as residual metal limits are easier to meet. The straightforward isolation procedure also facilitates the recycling of solvents, further enhancing the sustainability profile. These factors collectively make the technology attractive for long-term partnerships focused on environmentally responsible chemical production.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing a clear understanding of the process capabilities. Clients often seek clarification on catalyst loading, substrate scope, and purification requirements to assess feasibility for their specific projects. The answers below reflect the robust performance characteristics observed across multiple examples in the patent.
Q: What are the primary advantages of this copper-catalyzed method over traditional cross-coupling?
A: This method eliminates the need for expensive transition metal catalysts and pre-functionalized pyridine substrates, operating under mild conditions with high atom economy and broad substrate tolerance.
Q: Is this synthesis suitable for large-scale commercial production?
A: Yes, the process utilizes readily available raw materials, mild reaction temperatures (25-35°C), and simple post-processing extraction, making it highly scalable for industrial manufacturing.
Q: What is the typical purity profile of the resulting derivatives?
A: The reaction exhibits strong specificity and high yields, allowing for the production of high-purity intermediates after standard column chromatography purification, suitable for sensitive pharmaceutical applications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2,4-Diaryl-6-Trifluoromethylpyridine Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this copper-catalyzed synthesis for the production of high-value fluorinated pyridines. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from lab bench to market is seamless. Our state-of-the-art facilities are equipped to handle the specific solvent systems and mild thermal requirements of this process, guaranteeing stringent purity specifications for every batch. With rigorous QC labs and a commitment to process safety, we deliver intermediates that meet the highest international standards, supporting your pipeline from early development through commercial launch.
We invite you to leverage our technical expertise to optimize this route for your specific molecular targets. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are prepared to provide specific COA data and comprehensive route feasibility assessments to demonstrate how this innovative method can enhance your supply chain efficiency and reduce overall manufacturing costs. Let us collaborate to bring your next-generation therapeutics to market faster and more economically.