Revolutionizing Trichostatin A Production: A Scalable Chiral Synthesis Strategy for Global Pharma
Revolutionizing Trichostatin A Production: A Scalable Chiral Synthesis Strategy for Global Pharma
The pharmaceutical landscape for oncology therapeutics is constantly evolving, driven by the demand for more efficient and cost-effective manufacturing processes for critical active pharmaceutical ingredients (APIs). A pivotal advancement in this domain is detailed in patent CN1939898B, which outlines a groundbreaking chiral total synthesis method for Trichostatin A, a potent histone deacetylase (HDAC) inhibitor. This natural product, originally isolated from Streptomyces hygroscopicus, has demonstrated profound potential in inducing tumor cell differentiation and apoptosis, making it a cornerstone compound for cancer research and drug development. The disclosed methodology represents a significant departure from traditional synthetic approaches, leveraging organocatalysis to establish chirality with high precision while utilizing readily available commodity chemicals. For R&D directors and procurement specialists alike, this patent offers a compelling blueprint for optimizing the supply chain of complex chiral intermediates, promising enhanced stability, operational simplicity, and a marked improvement in overall process efficiency compared to legacy technologies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the innovations presented in this patent, the synthetic landscape for Trichostatin A was dominated by the route reported by Mori et al. in 1988, which, while pioneering, suffered from substantial logistical and economic drawbacks that hindered large-scale adoption. The conventional methodology relied heavily on expensive chiral pool starting materials, specifically methyl 3-hydroxy-2-methylpropionate, which not only inflated raw material costs but also introduced supply chain vulnerabilities associated with sourcing specialized fine chemicals. Furthermore, the intermediates generated throughout the Mori synthesis were characterized by poor stability, necessitating strict experimental controls, cryogenic conditions, and immediate processing that complicated warehouse management and increased the risk of batch failure. The cumulative effect of these inefficiencies was a prohibitively low overall yield of merely 6.1%, rendering the process economically unviable for commercial-scale manufacturing and limiting the availability of high-purity Trichostatin A for broader clinical and research applications.
The Novel Approach
In stark contrast, the novel synthesis route delineated in patent CN1939898B introduces a paradigm shift by employing an organocatalytic asymmetric aldol reaction as the foundational step, utilizing L-proline as a cheap and robust catalyst. This approach eliminates the dependency on costly chiral sources, substituting them with abundant feedstocks like p-nitrobenzaldehyde and propionaldehyde, which are accessible from global chemical suppliers at a fraction of the cost. The operational simplicity is further enhanced by the ability to conduct key reactions at room temperature, drastically reducing energy consumption and the need for specialized low-temperature reactor infrastructure. Most critically, the new route boasts intermediates with superior stability, allowing for more flexible processing windows and easier purification, which collectively contribute to a doubled overall yield of 12.9%. This substantial improvement in process metrics translates directly into a more reliable supply of high-purity pharmaceutical intermediates, addressing the critical pain points of cost and scalability faced by modern drug manufacturers.
Mechanistic Insights into L-Proline Catalyzed Asymmetric Aldol Reaction
The cornerstone of this synthetic breakthrough lies in the initial organocatalytic step, where L-proline facilitates the enantioselective coupling of p-nitrobenzaldehyde and propionaldehyde to form the chiral beta-hydroxy aldehyde intermediates 2a and 2b. Mechanistically, L-proline acts as a chiral amine catalyst, forming a transient enamine species with the propionaldehyde that lowers the activation energy for the nucleophilic attack on the aldehyde electrophile. The rigid pyrrolidine ring of the proline catalyst imposes a specific spatial arrangement on the transition state, effectively shielding one face of the enamine and directing the incoming aldehyde to approach from the less hindered side, thereby establishing the desired stereochemistry with high fidelity. This biomimetic strategy not only avoids the use of toxic heavy metals often found in traditional asymmetric catalysis but also operates under mild conditions that preserve the integrity of sensitive functional groups present in the substrate.
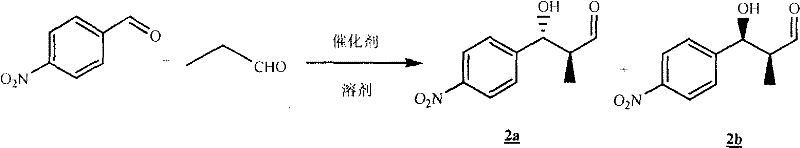
Following the establishment of the chiral center, the synthesis proceeds through a series of protection and elongation steps designed to maintain stereochemical integrity while building the complex polyene backbone characteristic of Trichostatin A. A crucial aspect of impurity control in this route is the strategic separation of diastereomers at the silyl protection stage, where compounds 4a and 4b are generated. Although the initial aldol reaction produces a mixture, the subsequent derivatization into silyl ethers creates distinct physical properties between the diastereomers, allowing for their complete separation via standard column chromatography. This purification checkpoint ensures that only the desired stereoisomer 4a proceeds down the synthetic pipeline, effectively scrubbing out unwanted enantiomeric impurities early in the process. By isolating the correct configuration before the resource-intensive chain elongation steps, the process minimizes the waste of reagents on incorrect isomers and guarantees that the final API meets the rigorous enantiomeric excess (ee) specifications required for biological activity.
How to Synthesize Trichostatin A Efficiently
The synthesis of Trichostatin A via this novel route involves a logical sequence of thirteen distinct chemical transformations that convert simple aromatic and aliphatic aldehydes into the complex hydroxamic acid structure. The process begins with the organocatalytic aldol condensation, followed by acetal protection to mask the aldehyde functionality, and silyl protection of the secondary alcohol to prevent side reactions during subsequent steps. Once the chiral core is secured and purified, the carbon chain is extended through iterative Wittig or Horner-Emmons olefinations, interspersed with selective reductions and oxidations to install the conjugated double bond system. The detailed standardized synthesis steps, including specific reagent stoichiometry, solvent choices, and reaction times for each of the thirteen stages, are outlined below to facilitate technology transfer and process validation.
- Construct the chiral core via L-proline catalyzed asymmetric aldol reaction between p-nitrobenzaldehyde and propionaldehyde, followed by acetal and silyl protection.
- Elongate the carbon chain through sequential Wittig or Horner-Emmons olefinations, interspersed with selective reduction and oxidation steps to establish the conjugated system.
- Finalize the molecule by reducing the nitro group, performing reductive amination, forming the hydroxamic acid, and executing the final benzylic oxidation.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthesis route offers tangible strategic advantages that extend beyond mere technical feasibility, directly impacting the bottom line and operational resilience. The shift from specialized chiral pool reagents to commodity chemicals like p-nitrobenzaldehyde and propionaldehyde significantly mitigates supply risk, as these materials are produced in massive volumes globally, ensuring consistent availability and price stability even during market fluctuations. Furthermore, the elimination of cryogenic requirements for the key stereoselective step reduces the capital expenditure needed for specialized reactor equipment, allowing for production in standard multipurpose facilities that are easier to validate and maintain. The improved stability of intermediates also simplifies logistics, reducing the need for expedited shipping or cold-chain storage between synthesis steps, which translates into lower freight costs and reduced carbon footprint for the manufacturing operation.
- Cost Reduction in Manufacturing: The economic viability of this process is driven by the drastic reduction in raw material costs and the doubling of overall yield from 6.1% to 12.9% compared to prior art. By replacing expensive chiral starting materials with inexpensive organocatalysts and commodity aldehydes, the direct material cost per kilogram of the final intermediate is substantially lowered. Additionally, the higher yield means that less solvent and reagent volume is required per unit of output, reducing waste disposal costs and improving the overall mass intensity of the process. The operational simplicity, characterized by room temperature reactions and standard workup procedures, further decreases labor and utility costs, making the commercial scale-up of complex chiral intermediates far more economically attractive.
- Enhanced Supply Chain Reliability: Reliability is bolstered by the use of robust, non-hazardous reagents such as L-proline and common silyl chlorides, which are widely stocked by chemical distributors worldwide, minimizing the risk of single-source bottlenecks. The stability of the protected intermediates, particularly the silyl ether 4a, allows for the potential creation of strategic stockpiles or semi-finished goods inventory, providing a buffer against demand spikes or temporary production interruptions. This flexibility enables a more responsive supply chain capable of meeting just-in-time delivery schedules for downstream API manufacturers without compromising on quality or lead times.
- Scalability and Environmental Compliance: From an environmental and scalability perspective, the avoidance of heavy metal catalysts and the use of greener solvents like ethanol or ethyl acetate in various steps align with modern green chemistry principles and regulatory expectations. The process generates less hazardous waste, simplifying effluent treatment and reducing the environmental compliance burden on the manufacturing site. The linear nature of the synthesis, combined with high-yielding individual steps, ensures that scaling from pilot plant to multi-ton commercial production is predictable and manageable, reducing the technical risk typically associated with introducing new chiral syntheses into the supply chain.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Trichostatin A synthesis route, derived from the specific advantages and mechanistic details disclosed in the patent literature. Understanding these nuances is essential for technical teams evaluating the feasibility of integrating this process into their existing manufacturing portfolios or for procurement specialists negotiating supply agreements. The answers provided reflect the empirical data and process parameters established in the patent, ensuring accuracy and reliability for decision-making purposes.
Q: How does the new synthesis route improve yield compared to conventional methods?
A: The novel route described in patent CN1939898B achieves a total yield of 12.9%, which is significantly higher than the 6.1% yield reported in the conventional Mori route, primarily due to improved intermediate stability and simpler operational conditions.
Q: What is the primary advantage of using L-proline in the first step?
A: L-proline serves as an inexpensive, metal-free organocatalyst that facilitates the asymmetric aldol reaction at room temperature, eliminating the need for costly chiral pool starting materials and harsh reaction conditions.
Q: How is optical purity ensured during the synthesis?
A: Optical purity is established early via the enantioselective aldol reaction and further refined through the chromatographic separation of diastereomeric intermediates (4a and 4b), ensuring the final product meets stringent stereochemical requirements.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trichostatin A Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of robust synthetic routes in securing the supply of life-saving oncology therapeutics. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. We are committed to delivering high-purity Trichostatin A and its key intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. By leveraging advanced chiral synthesis technologies like the organocatalytic route described herein, we provide our partners with a competitive edge through superior quality and consistent supply continuity.
We invite you to collaborate with us to optimize your supply chain for HDAC inhibitors and related pharmaceutical intermediates. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can support your drug development timelines and commercial launch goals.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →