Commercializing Next-Generation Wee1 Kinase Inhibitors: Scalable Synthesis of Dihydropyrazolopyrimidinone Derivatives
The global oncology therapeutic landscape is undergoing a paradigm shift towards targeted kinase inhibitors that can synergize with traditional chemotherapy and radiotherapy. Patent CN114591334A discloses a novel class of dihydropyrazolopyrimidinone derivatives designed as potent Wee1 kinase inhibitors, addressing the critical need for agents that can overcome DNA damage repair mechanisms in tumor cells. These compounds feature a unique structural architecture incorporating bridged or spiro-cyclic amine moieties linked to a pyrazolopyrimidinone core, offering enhanced selectivity and metabolic profiles. As a leading manufacturer, we recognize the immense potential of this chemical space for developing next-generation cancer therapies. The structural versatility allows for extensive SAR exploration while maintaining a scalable synthetic backbone suitable for industrial production.
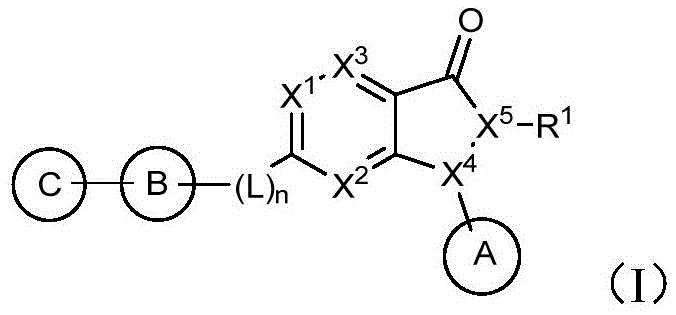
The development of high-purity pharmaceutical intermediates requires a deep understanding of the underlying chemical transformations. The disclosed technology provides a robust framework for synthesizing these complex heterocycles, ensuring that the final active pharmaceutical ingredients meet stringent regulatory standards. By focusing on the specific substitution patterns defined in the patent, particularly the variations in Ring C and the linker L, manufacturers can optimize potency against Wee1-mediated diseases. This technical insight report analyzes the synthetic feasibility and commercial advantages of adopting this novel scaffold for your oncology pipeline.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthesis routes for kinase inhibitors often rely heavily on palladium-catalyzed cross-coupling reactions for installing aryl amine substituents. While effective, these methods introduce significant challenges for large-scale manufacturing, including the removal of trace heavy metal residues to meet ppm-level specifications. Furthermore, conventional leaving groups such as chlorides often require harsh reaction conditions, elevated temperatures, or strong bases that can compromise sensitive functional groups elsewhere in the molecule. This frequently leads to lower overall yields and complex purification workflows, driving up the cost of goods and extending lead times for clinical supply. Additionally, many existing Wee1 inhibitors suffer from rapid metabolic clearance, necessitating frequent dosing regimens that impact patient compliance.
The Novel Approach
The methodology described in CN114591334A introduces a strategic innovation by utilizing a methylsulfinyl activation strategy for the final coupling step. Instead of relying solely on halide displacement, the process involves the oxidation of a methylthio precursor to a methylsulfinyl group, which serves as a superior leaving group. This transformation enables nucleophilic aromatic substitution to proceed under much milder conditions, typically at room temperature or with gentle heating. This approach not only improves the chemoselectivity of the reaction but also simplifies the downstream processing by reducing the formation of side products. The result is a cleaner reaction profile that facilitates easier isolation of the target dihydropyrazolopyrimidinone derivatives with high purity.
Mechanistic Insights into Methylsulfinyl Activation and Coupling
The core mechanistic advantage of this synthetic route lies in the electronic modulation of the pyrimidine ring. The initial formation of the pyrazolo[3,4-d]pyrimidin-3-one scaffold establishes the fundamental kinase inhibitor pharmacophore. Subsequent oxidation of the sulfur atom at the C-6 position using reagents like m-CPBA increases the electrophilicity of the adjacent carbon. This activation is crucial because it lowers the energy barrier for the nucleophilic attack by the bulky, sterically hindered bridged or spiro-cyclic anilines. Unlike traditional chloride leaving groups, the sulfinyl moiety allows the reaction to proceed efficiently without the need for aggressive catalysts that might degrade the sensitive allyl or hydroxyalkyl substituents present on the nitrogen atoms.
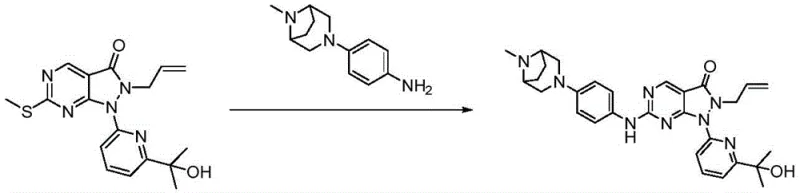
Furthermore, the impurity profile is significantly controlled through this mechanism. The mild conditions prevent the isomerization of the allyl group or the dehydration of the hydroxy-isopropyl moiety, which are common degradation pathways in harsher acidic or basic environments. The use of organic bases like DIPEA in the coupling step ensures that the reaction mixture remains homogeneous and manageable. From a quality control perspective, this translates to a more consistent batch-to-batch reproducibility. The ability to tolerate diverse amine structures, including the complex 3,8-diazabicyclo[3.2.1]octane and diazaspiro systems, demonstrates the robustness of this chemical platform for generating a wide library of analogs for preclinical evaluation.
How to Synthesize Dihydropyrazolopyrimidinone Efficiently
The synthesis of these Wee1 inhibitors follows a logical convergent strategy that separates the construction of the heterocyclic core from the installation of the complex amine side chains. This modularity is essential for process chemistry, as it allows for the optimization of each fragment independently before the final convergence. The process begins with the condensation of hydrazine derivatives with pyrimidine esters to form the fused ring system, followed by N-alkylation to introduce the allyl group. The critical activation step involves the selective oxidation of the thioether, which must be carefully monitored to prevent over-oxidation to the sulfone. Detailed standardized synthesis steps see the guide below.
- Construct the pyrazolo[3,4-d]pyrimidin-3-one core via cyclization of hydrazino-pyrimidine esters under basic conditions.
- Activate the C-6 position by oxidizing the methylthio group to a methylsulfinyl group using m-CPBA.
- Perform nucleophilic aromatic substitution with complex bridged or spiro-cyclic anilines to install the pharmacophore.
Commercial Advantages for Procurement and Supply Chain Teams
Adopting this synthetic route offers tangible benefits for procurement strategies focused on cost efficiency and supply security. The elimination of palladium catalysts in the final coupling step removes a major cost driver and a significant supply chain bottleneck associated with precious metal sourcing and recovery. This shift to organocatalytic or reagent-based activation reduces the dependency on volatile metal markets and simplifies the waste management protocols required for GMP manufacturing. Consequently, the overall cost of production is significantly reduced, making these advanced intermediates more accessible for early-stage drug development programs.
- Cost Reduction in Manufacturing: The replacement of transition metal-catalyzed couplings with a methylsulfinyl displacement strategy drastically lowers the raw material costs associated with ligands and metal salts. Moreover, the simplified purification requirements reduce solvent consumption and processing time, leading to substantial operational savings. By avoiding the need for specialized scavengers to remove heavy metals, the manufacturing process becomes leaner and more economically viable for commercial scale-up.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis, such as methyl picolinates and substituted hydrazines, are commodity chemicals with stable global supply chains. The robust nature of the reaction conditions means that production is less susceptible to disruptions caused by equipment limitations or strict environmental controls required for sensitive catalytic processes. This reliability ensures consistent delivery timelines for clinical trial materials and commercial batches.
- Scalability and Environmental Compliance: The synthetic pathway is designed with scalability in mind, utilizing common solvents like toluene, ethanol, and dichloromethane which are easily recovered and recycled. The absence of heavy metal waste streams simplifies environmental compliance and reduces the burden on wastewater treatment facilities. This green chemistry approach aligns with modern sustainability goals, making the manufacturing process more attractive for long-term partnerships and regulatory approval.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these dihydropyrazolopyrimidinone derivatives. Understanding these aspects is vital for integrating this technology into your existing drug discovery and development workflows. The answers are derived directly from the technical specifications and experimental data provided in the patent documentation.
Q: What is the key advantage of the methylsulfinyl activation strategy in this synthesis?
A: The oxidation of the methylthio group to methylsulfinyl creates a highly reactive leaving group that facilitates nucleophilic substitution under mild conditions, eliminating the need for expensive transition metal catalysts in the final coupling step.
Q: How does the metabolic stability of these derivatives compare to existing inhibitors like AZD1775?
A: Experimental data indicates that specific compounds within this class demonstrate significantly improved metabolic stability in human, monkey, and dog liver microsomes compared to the reference compound AZD1775, suggesting better in vivo exposure.
Q: Are the starting materials for the bridged amine moieties commercially viable?
A: Yes, the synthesis utilizes readily available precursors such as diazabicyclo and diazaspiro scaffolds, which can be sourced or synthesized at scale, ensuring a robust supply chain for commercial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Dihydropyrazolopyrimidinone Supplier
The technical potential of these Wee1 kinase inhibitors is matched by our capability to deliver them at scale. NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met regardless of the project phase. Our facility is equipped with rigorous QC labs and stringent purity specifications to guarantee that every batch of intermediate meets the highest industry standards. We understand the critical nature of oncology APIs and are committed to supporting your timeline with reliable manufacturing execution.
We invite you to discuss how our expertise can optimize your supply chain for these novel compounds. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how we can add value to your project immediately.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →