Advanced Macrocyclic Tyrosine Kinase Inhibitors: Scalable Synthesis for Next-Gen Oncology APIs
Advanced Macrocyclic Tyrosine Kinase Inhibitors: Scalable Synthesis for Next-Gen Oncology APIs
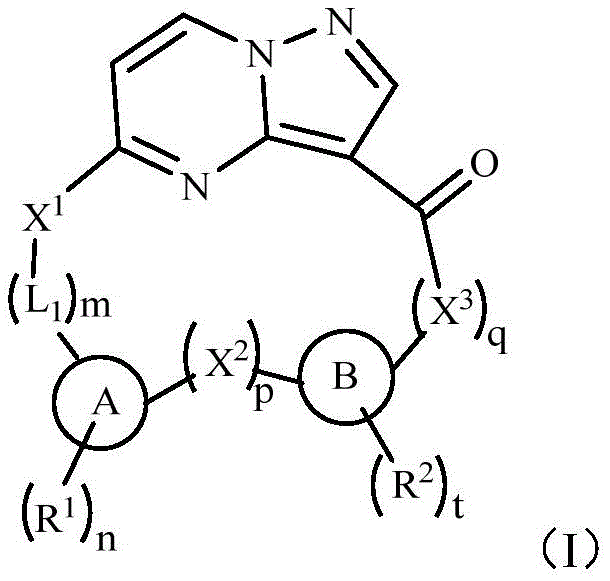
The landscape of oncology drug development is rapidly shifting towards highly selective small molecules capable of overcoming acquired resistance mechanisms. Patent CN111171049A discloses a groundbreaking class of macrocyclic tyrosine kinase inhibitors targeting TRK, ALK, and ROS1 receptors, which are critical drivers in various solid tumors. These novel compounds, characterized by a pyrazolo[1,5-a]pyrimidine core fused with complex heterocyclic systems, represent a significant leap forward in treating cancers that have developed resistance to first-generation therapies. For pharmaceutical manufacturers and procurement leaders, understanding the synthetic accessibility and structural nuances of these intermediates is paramount for securing a reliable supply chain for next-generation anticancer agents. The detailed chemical architecture presented in this patent offers a robust blueprint for the industrial production of high-purity active pharmaceutical ingredients (APIs) tailored for precision medicine applications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional tyrosine kinase inhibitors often suffer from limited selectivity and rapid onset of drug resistance due to point mutations in the kinase domain, such as the gatekeeper mutations found in NTRK and ALK genes. Conventional linear inhibitors typically rely on flexible linkers that entropic penalties reduce binding affinity, necessitating higher dosages that increase off-target toxicity. Furthermore, the synthesis of earlier generation kinase inhibitors frequently involves harsh reaction conditions, expensive transition metal catalysts that require rigorous removal to meet regulatory standards, and protection group strategies that add unnecessary steps and reduce overall atom economy. These inefficiencies translate directly into higher manufacturing costs and extended lead times for clinical trial materials, creating bottlenecks for reliable pharmaceutical intermediates supplier networks trying to meet the urgent demands of oncology research.
The Novel Approach
The innovative strategy outlined in the patent employs a macrocyclic scaffold that constrains the molecular conformation, effectively reducing the entropic cost of binding and significantly enhancing potency against resistant mutants. By integrating fused ring systems like benzofuran or cyclopenta[c]pyridine moieties directly into the macrocycle, the design achieves superior pharmacokinetic profiles and metabolic stability compared to acyclic analogs. This structural rigidity allows for precise positioning of pharmacophores within the ATP-binding pocket, ensuring sustained inhibition even in the presence of common resistance mutations like G595R. From a process chemistry perspective, this approach utilizes robust carbon-carbon and carbon-nitrogen bond-forming reactions that are well-suited for large-scale optimization, facilitating cost reduction in API manufacturing by minimizing purification burdens and maximizing yield consistency across batches.
Mechanistic Insights into Macrocyclization and Heterocycle Formation
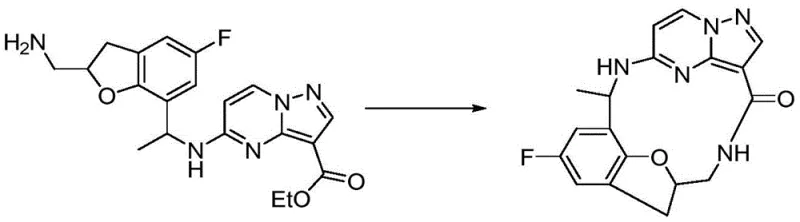
The core synthetic challenge lies in the construction of the macrocyclic ring system, which typically involves a late-stage intramolecular amide coupling or nucleophilic substitution to close the ring. As illustrated in the synthesis of Compound 17, the process begins with the functionalization of a fluorophenol precursor, where regioselective bromination and allylation set the stage for the formation of the dihydrobenzofuran core. This intermediate is then elaborated through a series of protective group manipulations, including phthalimide installation and subsequent reductive amination, to introduce the necessary amine functionality for coupling with the pyrazolo[1,5-a]pyrimidine electrophile. The careful selection of reagents such as lithium hexamethyldisilazide (LiHMDS) for enolate generation and fluorodiphenylphosphine oxide (FDPP) for peptide-like coupling ensures high fidelity in bond formation while minimizing racemization of chiral centers.
Impurity control is rigorously managed through the use of crystalline intermediates and chromatographic purification at critical junctures, particularly before the final macrocyclization step where intermolecular oligomerization is a competing side reaction. The patent details specific conditions, such as microwave-assisted heating for certain transformations, which accelerate reaction kinetics and improve throughput without compromising product quality. By employing orthogonal protecting groups like Boc and phthalimide, the synthesis allows for selective deprotection sequences that reveal reactive amines only when needed, thereby preventing premature side reactions. This level of mechanistic control is essential for producing high-purity pharmaceutical intermediates that meet the stringent specifications required for global regulatory submissions and clinical safety.
How to Synthesize Compound 17 Efficiently
The synthesis of Compound 17 serves as a prime example of the platform technology described in the patent, demonstrating a convergent route that joins two complex fragments prior to the final ring-closing event. The process initiates with the preparation of a functionalized benzofuran amine, which is coupled to an ethyl pyrazolo[1,5-a]pyrimidine-3-carboxylate derivative under basic conditions to form the linear precursor. Following hydrolysis of the ester and removal of the phthalimide protecting group, the resulting amino acid undergoes intramolecular cyclization to yield the target macrocycle. Detailed standardized synthetic steps for this transformation are provided in the guide below to assist process chemists in replicating this high-value pathway.
- Preparation of the benzofuran core via allylation and subsequent cyclization of fluorophenol derivatives.
- Introduction of the amine side chain through phthalimide protection and reductive amination with acetyl groups.
- Coupling with the pyrazolo[1,5-a]pyrimidine ester followed by deprotection and final macrocyclization using FDPP.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers distinct strategic advantages centered around raw material availability and process robustness. The starting materials, such as fluorophenols and substituted pyrimidines, are commodity chemicals available from multiple global sources, mitigating the risk of single-supplier dependency that often plagues specialized oncology programs. Furthermore, the elimination of exotic catalysts in favor of standard organic reagents simplifies the waste management profile and reduces the environmental compliance burden associated with heavy metal disposal. This streamlined approach ensures enhanced supply chain reliability, allowing manufacturers to maintain continuous production schedules even during periods of raw material volatility.
- Cost Reduction in Manufacturing: The synthetic pathway leverages high-yielding transformations and avoids the use of precious metal catalysts like palladium or platinum in the final steps, which significantly lowers the cost of goods sold (COGS). By utilizing efficient coupling reagents and avoiding cryogenic conditions for the majority of the sequence, energy consumption is minimized, contributing to substantial cost savings in pharmaceutical intermediates manufacturing. Additionally, the ability to purify intermediates via crystallization rather than preparative HPLC reduces solvent usage and processing time, further driving down operational expenses.
- Enhanced Supply Chain Reliability: The modular nature of the synthesis allows for the parallel production of key fragments, such as the benzofuran amine and the pyrimidine acid, which can be stockpiled independently to buffer against supply disruptions. This flexibility ensures reducing lead time for high-purity pharmaceutical intermediates, enabling faster response to clinical trial demands. The robustness of the chemical steps means that technology transfer between manufacturing sites is straightforward, securing the continuity of supply for critical cancer therapies.
- Scalability and Environmental Compliance: The process is designed with commercial scale-up of complex macrocycles in mind, utilizing solvents and reagents that are compatible with large-scale reactor infrastructure. The absence of genotoxic impurities in the final steps and the use of greener alternatives for oxidation and reduction reactions align with modern environmental, social, and governance (ESG) goals. This commitment to sustainable chemistry not only facilitates regulatory approval but also enhances the corporate reputation of partners involved in the production of these life-saving medicines.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these macrocyclic kinase inhibitors. These insights are derived directly from the experimental data and structural analysis provided in the patent documentation, offering clarity for R&D and procurement stakeholders evaluating this technology for their pipelines.
Q: What specific mutations do these macrocyclic inhibitors target?
A: These compounds are designed to inhibit TRK, ALK, and ROS1 kinases, specifically addressing drug-resistant mutations such as NTRK1-G595R and NTRK3-G623R which render first-generation inhibitors ineffective.
Q: How does the macrocyclic structure improve efficacy?
A: The macrocyclic constraint pre-organizes the molecule into a bioactive conformation, enhancing binding affinity and selectivity for the ATP-binding pocket of the kinase domain while improving metabolic stability.
Q: Is the synthesis route scalable for commercial production?
A: Yes, the described methodology utilizes standard organic transformations such as nucleophilic substitution, reductive amination, and amide coupling, which are amenable to scale-up from kilogram to multi-ton production levels.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Compound 17 Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of securing a stable supply of advanced oncology intermediates to support the development of next-generation cancer therapies. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that complex macrocyclic structures like Compound 17 can be manufactured with consistent quality and efficiency. We operate stringent purity specifications and maintain rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify the identity and potency of every batch, guaranteeing that our clients receive materials that meet the highest industry standards for clinical and commercial use.
We invite you to collaborate with us to optimize your supply chain for these vital tyrosine kinase inhibitors. Contact our technical procurement team today to request a Customized Cost-Saving Analysis for your specific project needs. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can accelerate your drug development timeline and reduce overall project costs.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →