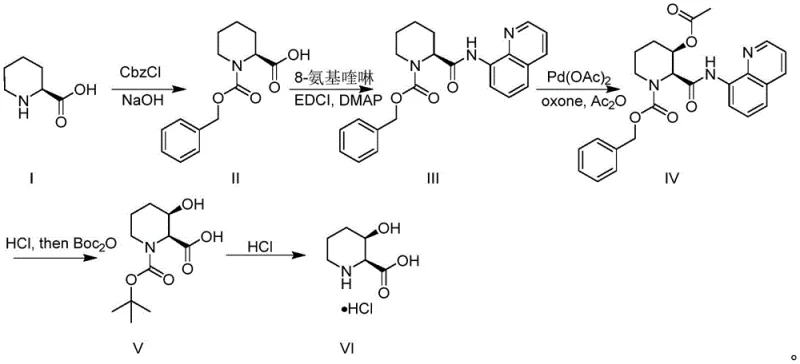
Scalable Synthesis of (2S,3R)-3-Hydroxy-2-Piperidinecarboxylic Acid via Pd-Catalysis
Introduction to Advanced Amino Acid Synthesis
The landscape of pharmaceutical intermediate manufacturing is constantly evolving, driven by the need for more efficient and sustainable synthetic routes. A pivotal development in this field is documented in patent CN112851568B, which outlines a robust method for synthesizing (2S,3R)-3-hydroxy-2-piperidinecarboxylic acid and its hydrochloride salt. This specific chiral building block is a critical structural motif found in numerous bioactive natural products, including the antitumor antibiotic tetrazomine and the antibacterial agent GE81112A. Historically, accessing these complex scaffolds has been a bottleneck due to lengthy synthetic sequences and poor stereocontrol. The disclosed technology represents a significant leap forward, leveraging 8-aminoquinoline-directed palladium catalysis to achieve high diastereoselectivity in a streamlined fashion. For R&D directors and procurement specialists alike, understanding this methodology is crucial for securing reliable supply chains for next-generation therapeutics.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this innovation, the synthesis of (2S,3R)-3-hydroxy-2-piperidinecarboxylic acid was fraught with inefficiencies that hindered commercial viability. Traditional approaches often relied on multi-step syntheses starting from chiral pools like D-serine or D-glucose, which could entail upwards of 11 to 15 reaction steps with cumulative yields as low as 24% to 27%. Alternatively, asymmetric reactions constructing two chiral centers simultaneously often required complex organocatalysts and still resulted in long routes exceeding 12 steps. Enzymatic resolution methods, while capable of achieving optical purity, suffered from significant operational drawbacks, including reaction times extending up to 3 days and the necessity for cumbersome protection group strategies. Furthermore, earlier palladium-catalyzed attempts utilizing iodobenzene diacetate as an oxidant were limited to milligram scales and faced severe yield losses upon scaling due to difficult purification of iodobenzene by-products.
The Novel Approach
The methodology presented in the patent data offers a transformative solution by drastically reducing the step count and simplifying the operational complexity. By utilizing commercially available and inexpensive (S)-piperidine-2-carboxylic acid as the starting material, the route bypasses the need for expensive chiral pool precursors. The core innovation lies in the installation of an 8-aminoquinoline directing group, which facilitates a highly selective palladium-catalyzed C-H acetoxylation. This single step effectively constructs the critical hydroxyl stereocenter with excellent diastereocontrol. Unlike previous methods requiring multiple chromatographic purifications, this entire sequence necessitates only one column chromatography step, specifically after the acetoxylation reaction. The subsequent deprotection and salt formation steps are straightforward acid-base manipulations, making the process inherently safer and more amenable to industrial scale-up compared to the fragile enzymatic or long linear syntheses of the past.
Mechanistic Insights into 8-Aminoquinoline Directed C-H Activation
The heart of this synthetic strategy is the palladium-catalyzed acetoxylation step, which exemplifies modern C-H functionalization chemistry. The mechanism initiates with the coordination of the palladium catalyst, typically palladium acetate, to the nitrogen atoms of the 8-aminoquinoline amide substrate. This bidentate coordination forms a stable five-membered palladacycle intermediate, effectively positioning the metal center in close proximity to the C3-H bond of the piperidine ring. This geometric constraint is what drives the remarkable regioselectivity, ensuring activation occurs exclusively at the beta-position relative to the carbonyl. Once the C-H bond is activated, the system undergoes oxidation in the presence of Oxone (potassium peroxymonosulfate) and acetic anhydride. This oxidative environment likely generates a high-valent palladium species or facilitates a Pd(II)/Pd(IV) cycle that allows for the insertion of the acetoxy group. The use of acetic anhydride serves a dual purpose as both a solvent component and the source of the acetyl group, which is later hydrolyzed to reveal the free hydroxyl functionality.
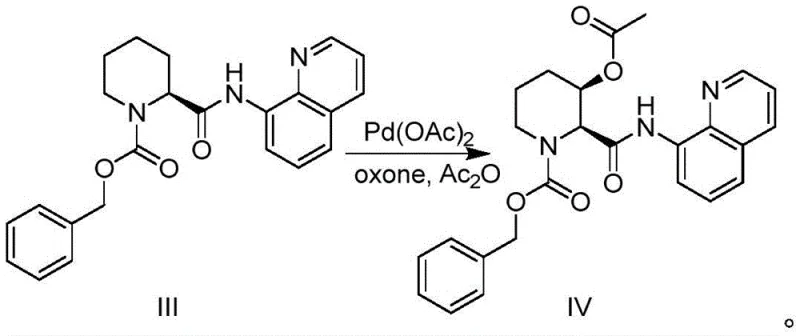
Controlling impurities in such transition metal-catalyzed reactions is paramount for pharmaceutical applications. A key advantage of this specific protocol is the choice of oxidant. Traditional methods employing iodobenzene diacetate generate stoichiometric amounts of iodobenzene, a lipophilic by-product that is notoriously difficult to remove and can participate in unwanted arylation side reactions with the substrate. In contrast, the use of Oxone generates inorganic sulfate salts which are water-soluble and can be easily removed during the aqueous workup. Additionally, the absence of copper co-catalysts, which were required in some prior art methods, eliminates the risk of heavy metal contamination that often necessitates expensive scavenging resins. The high diastereoselectivity observed, yielding the (2S,3R) isomer predominantly, suggests that the rigid palladacycle intermediate imposes a strict conformational bias during the bond-forming event, minimizing the formation of unwanted diastereomers and simplifying downstream purification efforts significantly.
How to Synthesize (2S,3R)-3-Hydroxy-2-Piperidinecarboxylic Acid Efficiently
Implementing this synthesis requires careful attention to the preparation of the directing group precursor and the optimization of the oxidation conditions. The process begins with the protection of the starting amino acid, followed by amide coupling to install the quinoline moiety. The critical acetoxylation step demands precise control of temperature and stoichiometry to maximize the conversion to the acetoxy intermediate. Following this, a tandem hydrolysis and protection sequence converts the intermediate into a stable Boc-protected hydroxy acid, which serves as a versatile handle for further derivatization or final salt formation. The detailed standardized synthesis steps are outlined in the guide below, providing a clear roadmap for laboratory execution and process validation.
- Protect (S)-piperidine-2-carboxylic acid with Cbz-Cl under basic conditions to form the N-Cbz protected intermediate.
- Couple the protected acid with 8-aminoquinoline using EDCI and DMAP to install the directing group.
- Perform Pd-catalyzed acetoxylation using Pd(OAc)2 and Oxone to introduce the hydroxyl group with high diastereoselectivity.
- Hydrolyze the amide and acetyl groups under acidic conditions, followed by Boc protection to isolate the key intermediate.
- Remove the Boc protecting group with HCl to yield the final (2S,3R)-3-hydroxy-2-piperidinecarboxylic acid hydrochloride.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this novel synthetic route offers tangible benefits that extend beyond mere technical elegance. The primary driver for cost reduction lies in the raw material selection and reagent efficiency. By shifting from expensive chiral sugars or amino acids to bulk-available (S)-pipecolic acid, the baseline material cost is significantly lowered. Moreover, the replacement of iodobenzene diacetate with Oxone represents a substantial economic improvement, as the patent data indicates Oxone is approximately three times less expensive on a weight basis. This substitution not only lowers the bill of materials but also reduces the waste disposal costs associated with iodinated organic by-products. The elimination of copper salts further reduces the burden on wastewater treatment facilities and lowers the cost of metal testing and clearance in the final API.
- Cost Reduction in Manufacturing: The streamlined nature of this synthesis directly translates to lower manufacturing costs through reduced labor and utility consumption. With a total step count that is significantly lower than the 11 to 15 steps required by traditional methods, the cumulative loss of material at each stage is minimized, leading to a higher overall yield. The fact that the entire route requires only one column chromatography step is a massive operational saving, as chromatography is often the most time-consuming and solvent-intensive unit operation in fine chemical synthesis. By avoiding multiple purifications, the consumption of silica gel and organic solvents is drastically curtailed, aligning with green chemistry principles and reducing the environmental footprint of the production process.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the use of commodity chemicals that are widely available from multiple global vendors. Reagents such as palladium acetate, acetic anhydride, and di-tert-butyl dicarbonate are standard inventory items for most contract manufacturing organizations, reducing the risk of single-source bottlenecks. The robustness of the reaction conditions, which tolerate a range of temperatures and do not require cryogenic conditions or inert atmospheres for every step, ensures that production can proceed with minimal downtime. This reliability is critical for maintaining continuous supply to downstream API manufacturers who depend on consistent delivery of high-quality intermediates to meet their own regulatory filing timelines.
- Scalability and Environmental Compliance: The process has demonstrated successful translation from milligram to gram scales with improved yields, indicating strong potential for kilogram and tonne-scale production. The comparative data shows that while older methods suffered yield drops upon scaling, this Pd-catalyzed route maintained a robust 67% yield at the gram scale. From an environmental compliance perspective, the avoidance of halogenated oxidants and copper catalysts simplifies the effluent profile. The aqueous waste streams primarily contain inorganic salts and organic acids which are easier to treat biologically or chemically compared to heavy metal-laden waste. This ease of waste management facilitates faster regulatory approvals for new manufacturing sites and reduces the long-term liability associated with hazardous waste storage and disposal.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on the practical aspects of adopting this technology. Understanding these nuances helps stakeholders make informed decisions about integrating this route into their existing manufacturing portfolios.
Q: What are the advantages of using Oxone over PhI(OAc)2 in this synthesis?
A: According to patent CN112851568B, Oxone is significantly more cost-effective, being approximately three times cheaper than iodobenzene diacetate. Furthermore, Oxone simplifies purification as it can be removed by filtration, whereas PhI(OAc)2 generates iodobenzene by-products that are difficult to separate and can lead to arylation side reactions.
Q: How does this method improve upon traditional enzymatic resolution?
A: Traditional enzymatic resolution methods often require extended reaction times, typically around 3 days, and involve tedious protection group manipulations. The novel palladium-catalyzed route described achieves high optical activity with fewer steps and eliminates the need for prolonged enzymatic incubation, thereby enhancing throughput.
Q: Is this synthesis suitable for large-scale manufacturing?
A: Yes, the process is designed for scalability. It utilizes inexpensive starting materials like (S)-pipecolic acid and requires only one column chromatography step throughout the entire sequence. Comparative data indicates superior yields at the gram scale compared to prior art methods using copper co-catalysts.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Hydroxy-2-Piperidinecarboxylic Acid Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient synthetic routes like the one described in CN112851568B for the production of complex pharmaceutical intermediates. Our team of process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to pilot plant is seamless. We are committed to delivering high-purity 3-hydroxy-2-piperidinecarboxylic acid derivatives that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our expertise in palladium catalysis and C-H activation allows us to optimize these reactions further, potentially improving yields and reducing catalyst loading even beyond the patent examples.
We invite you to collaborate with us to leverage this advanced technology for your drug development programs. Whether you require custom synthesis of specific analogs or large-scale supply of the parent compound, our technical procurement team is ready to assist. Please contact us to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage potential partners to reach out for specific COA data and route feasibility assessments to ensure this technology aligns perfectly with your project goals and timelines.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →