Advanced Manufacturing of 2-Aryloxy-4-Chloropyridines for Neurological Drug Development
Advanced Manufacturing of 2-Aryloxy-4-Chloropyridines for Neurological Drug Development
The pharmaceutical industry's relentless pursuit of novel therapeutics for neurological disorders has placed a spotlight on Corticotropin-Releasing Factor (CRF) antagonists, a class of compounds showing immense promise in treating anxiety, depression, and stress-related pathologies. At the heart of synthesizing these complex bioactive molecules lies a critical structural motif: the 2-aryloxy-4-chloropyridine scaffold. Patent CN1082949C discloses a highly specialized and efficient process for converting 2,4-dichloropyridine derivatives into these valuable intermediates. This technology represents a significant leap forward in heterocyclic chemistry, offering a robust pathway to access pharmacologically active 2-phenoxypyridine derivatives. By leveraging copper-catalyzed nucleophilic aromatic substitution, this method overcomes traditional selectivity challenges, ensuring that the reactive 4-chloro position remains available for subsequent diversification while successfully installing the aryl ether at the 2-position. For R&D directors and procurement strategists alike, understanding the nuances of this patented route is essential for securing a reliable supply chain of high-purity pharmaceutical intermediates.
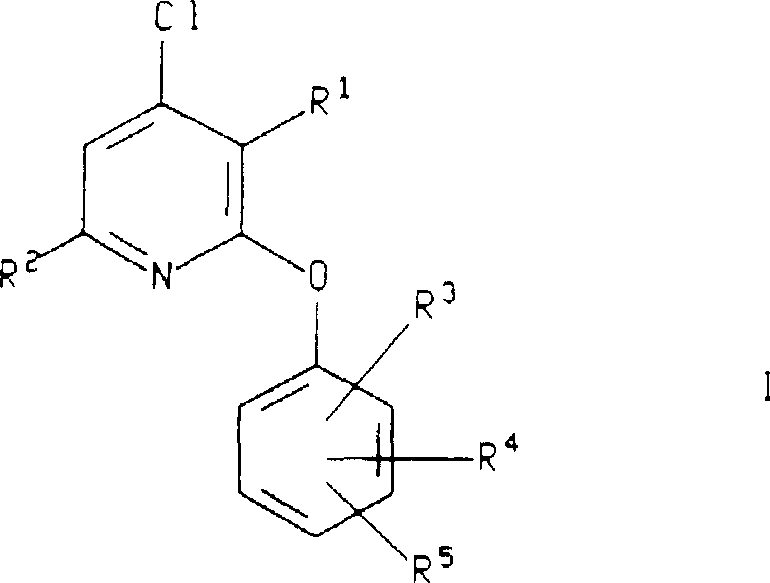
The versatility of this synthetic approach cannot be overstated, as it accommodates a wide range of substituents on both the pyridine ring and the phenolic partner. As illustrated in the general structure, the process allows for the independent variation of alkyl and alkoxy groups, enabling medicinal chemists to rapidly generate libraries of analogs for Structure-Activity Relationship (SAR) studies. This flexibility is paramount in the early stages of drug discovery, where minor structural modifications can drastically alter bioavailability and receptor binding affinity. Furthermore, the ability to synthesize these compounds as pharmaceutically acceptable salts adds another layer of utility, facilitating formulation development and stability testing. For companies aiming to establish themselves as a reliable pharmaceutical intermediate supplier, mastering this specific transformation provides a competitive edge in the crowded landscape of neurology drug development.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of unsymmetrical diaryl ethers or aryl-heteroaryl ethers has been plagued by significant chemical hurdles, primarily revolving around regioselectivity and reaction conditions. Traditional nucleophilic aromatic substitution (SNAr) on dichloropyridines often suffers from poor discrimination between the 2- and 4-positions, leading to complex mixtures of isomers that are notoriously difficult and costly to separate. In many legacy processes, harsh reaction conditions involving high temperatures and strong bases were required to drive the reaction, which frequently resulted in the degradation of sensitive functional groups or the formation of tar-like byproducts. Moreover, without a suitable catalyst, the reaction kinetics are often sluggish, requiring extended reaction times that reduce throughput and increase energy consumption. These inefficiencies translate directly into higher manufacturing costs and longer lead times, creating bottlenecks for procurement managers tasked with maintaining continuous supply for clinical trials. The inability to consistently preserve the 4-chloro handle for downstream coupling further limits the utility of intermediates produced via these older, less sophisticated methods.
The Novel Approach
The methodology outlined in CN1082949C introduces a paradigm shift by incorporating organometallic halides or oxides, specifically copper species, to mediate the coupling reaction. This copper-catalyzed approach dramatically enhances the nucleophilicity of the phenoxide ion towards the 2-position of the pyridine ring, effectively suppressing competing reactions at the 4-position. By operating under milder conditions—typically ranging from room temperature to 150°C—the process preserves the integrity of the substrate while achieving superior conversion rates. The use of solvents like pyridine or DMSO not only solubilizes the reactants but also stabilizes the catalytic species, ensuring a smooth reaction profile. This novel approach eliminates the need for exotic or prohibitively expensive catalysts, relying instead on abundant copper salts like cuprous iodide. For supply chain heads, this translates to a more resilient manufacturing process that is less susceptible to raw material shortages. The result is a streamlined synthesis that delivers high-purity intermediates with minimal impurity profiles, directly addressing the stringent quality requirements of modern API manufacturing.
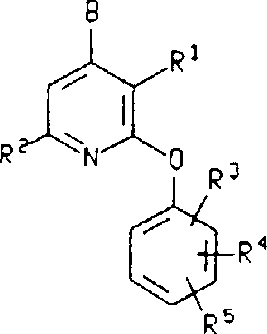
Mechanistic Insights into Copper-Catalyzed Etherification
To fully appreciate the technical superiority of this process, one must delve into the mechanistic underpinnings of the copper-catalyzed nucleophilic substitution. The reaction likely proceeds through a coordination complex where the copper center activates the aryl halide bond, facilitating the oxidative addition of the phenoxide nucleophile. This catalytic cycle lowers the activation energy barrier significantly compared to uncatalyzed thermal pathways. The presence of the base, such as potassium tert-butoxide or sodium hydride, is critical for generating the reactive phenoxide anion in situ from the corresponding phenol precursor. The choice of base and solvent system creates a synergistic effect; for instance, pyridine can act as both a solvent and a ligand, stabilizing the copper intermediate and preventing catalyst deactivation. This intricate balance ensures that the reaction proceeds with high fidelity, minimizing the formation of homocoupling byproducts or hydrolysis products. Understanding these mechanistic details allows process chemists to fine-tune reaction parameters, optimizing yield and purity for commercial scale-up of complex pharmaceutical intermediates.
Impurity control is another cornerstone of this patented technology, particularly concerning the preservation of the 4-chloro substituent. In the absence of the copper catalyst, there is a heightened risk of double substitution, where the phenol attacks both chloro positions, rendering the molecule useless for further elaboration into CRF antagonists. The catalytic system described herein exhibits remarkable chemoselectivity, preferentially targeting the 2-position due to electronic and steric factors enhanced by the metal center. Furthermore, the workup procedure detailed in the patent examples—involving sequential washes with ammonium hydroxide, sodium hydroxide, and hydrochloric acid—is designed to effectively remove residual copper species and unreacted starting materials. This rigorous purification protocol ensures that the final isolate meets the stringent purity specifications required for GMP production. For R&D teams, this level of control over the impurity profile reduces the burden on downstream purification steps, accelerating the overall development timeline.
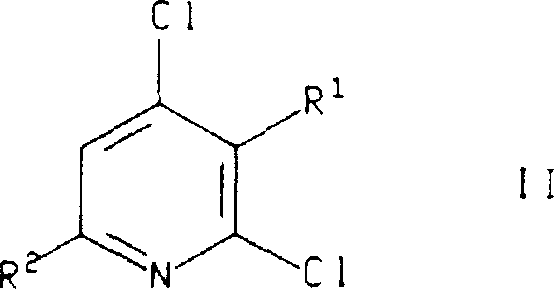
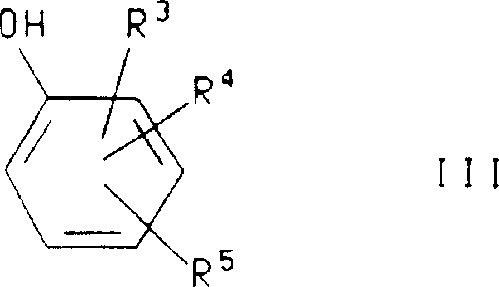
How to Synthesize 4-Chloro-3,6-dimethyl-2-(2,4,6-trimethylphenoxy)pyridine Efficiently
The practical execution of this synthesis is exemplified by the preparation of 4-chloro-3,6-dimethyl-2-(2,4,6-trimethylphenoxy)pyridine, a key building block for potent CRF antagonists. The process begins with the careful preparation of the reaction mixture in a dry, inert atmosphere to prevent moisture interference, which could deactivate the base or catalyst. The selection of reagents is precise: 2,4,6-trimethylphenol serves as the nucleophile, while 2,4-dichloro-3,6-lutidine acts as the electrophilic scaffold. The use of cuprous iodide as the catalyst and potassium tert-butoxide as the base in pyridine solvent creates an optimal environment for the coupling to occur. Following the reflux period, the quenching and extraction steps are critical for isolating the product in high yield, as demonstrated by the patent's reported isolation of the target compound as an off-white solid. The detailed standardized synthesis steps see the guide below.
- Prepare the reaction vessel with pyridine solvent and cool in an ice bath before adding 2,4,6-trimethylphenol and potassium tert-butoxide base.
- Introduce 2,4-dichloro-3,6-lutidine and cuprous iodide catalyst to the mixture, then heat to reflux for approximately 2 hours to drive the coupling.
- Quench the reaction with saturated ammonium chloride, perform extensive aqueous workup with acid and base washes, and crystallize the product from methanol.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this patented process offers substantial strategic benefits for organizations focused on cost reduction in API manufacturing. The reliance on copper catalysts rather than precious metals like palladium or platinum significantly lowers the raw material costs associated with the synthesis. Copper is abundant, inexpensive, and easier to source globally, mitigating the risks associated with supply chain volatility often seen with noble metals. Additionally, the process avoids the use of highly toxic or regulated reagents that would necessitate expensive waste disposal protocols, thereby reducing the environmental compliance burden. The robustness of the reaction conditions allows for operation in standard stainless steel reactors without the need for specialized glass-lined equipment, further capitalizing on existing infrastructure. These factors combine to create a manufacturing route that is not only chemically efficient but also economically sustainable, providing a clear pathway to substantial cost savings without compromising on quality.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the use of commodity chemicals like pyridine and simple alkali bases drive down the Bill of Materials (BOM) cost significantly. By avoiding complex protection-deprotection sequences often required in alternative routes, the overall step count is minimized, which directly correlates to reduced labor and utility costs. The high selectivity of the reaction minimizes the loss of valuable starting materials to byproducts, maximizing atom economy and ensuring that every kilogram of input contributes to the final output. This efficiency is crucial for maintaining healthy margins in the competitive generic and specialty chemical markets.
- Enhanced Supply Chain Reliability: The reagents specified in this process, including 2,4-dichloropyridines and substituted phenols, are widely available from multiple global suppliers, reducing dependency on single-source vendors. The stability of the intermediates and the final product allows for flexible inventory management, enabling manufacturers to produce in larger batches and store safely for extended periods. This reliability is vital for ensuring reducing lead time for high-purity heterocyclic compounds, allowing pharmaceutical partners to meet tight clinical trial deadlines without the fear of production delays. The scalability of the process ensures that supply can be ramped up quickly to meet surging demand as a drug candidate progresses through development phases.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated effectively in multi-gram to kilogram ranges within the patent literature, indicating readiness for ton-scale production. The solvent systems used, such as pyridine and methanol, are well-understood in terms of recovery and recycling, supporting green chemistry initiatives and reducing the carbon footprint of the manufacturing operation. The aqueous workup generates waste streams that are manageable with standard treatment facilities, avoiding the generation of hazardous heavy metal waste associated with other coupling methodologies. This alignment with environmental standards facilitates smoother regulatory approvals and enhances the corporate sustainability profile of the manufacturing entity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy and relevance for industry professionals. Understanding these details is crucial for making informed decisions about process adoption and supplier qualification. We encourage potential partners to review these insights carefully to gauge the fit for their specific project requirements.
Q: What is the primary advantage of using copper catalysis in this pyridine substitution?
A: The use of cuprous halides (like CuI) significantly enhances the regioselectivity of the nucleophilic attack at the 2-position of the pyridine ring, preventing unwanted disubstitution and ensuring the 4-chloro group remains intact for downstream functionalization.
Q: Can this process be scaled for commercial API production?
A: Yes, the process utilizes robust reagents like pyridine and potassium tert-butoxide which are readily available at industrial scales, and the workup involves standard extraction and crystallization techniques suitable for large-scale manufacturing.
Q: What are the critical impurities to monitor during this synthesis?
A: Key impurities include the 4-aryloxy isomer (if selectivity fails) and the di-aryloxy byproduct where both chloro groups are substituted; rigorous QC testing via HPLC is required to ensure the 4-chloro functionality is preserved.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Aryloxy-4-Chloropyridines Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the successful development of life-saving neurological therapies. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from the laboratory bench to full-scale manufacturing. We adhere to stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of 2-aryloxy-4-chloropyridines meets the highest industry standards. Our commitment to excellence extends beyond mere compliance; we strive to be a true partner in your drug development journey, offering technical support and process optimization services that add tangible value to your supply chain.
We invite you to contact our technical procurement team to discuss your specific requirements and explore how our capabilities align with your project goals. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into how our optimized manufacturing processes can reduce your overall project costs. We are ready to provide specific COA data and route feasibility assessments to demonstrate our competence and reliability. Let us collaborate to accelerate the delivery of innovative CRF antagonists to patients who need them most, leveraging our expertise in complex heterocyclic synthesis to drive your success.