Advanced Silicon-Mediated Synthesis for High-Purity Beta-Lactamase Inhibitor Intermediates
Introduction to Novel Intermediate Synthesis
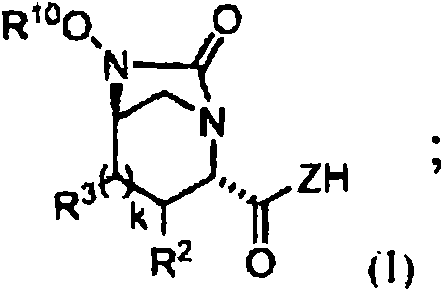
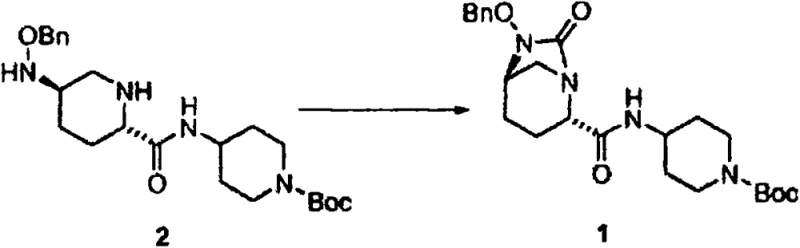
The pharmaceutical industry continuously seeks robust and scalable pathways for complex antibiotic intermediates, particularly those serving as key building blocks for beta-lactamase inhibitors. Patent CN107428784B discloses a groundbreaking process for the preparation of tert-butyl 4-((2S,5R)-6-(benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboxamido)piperidine-1-carboxylate and its analogues. This specific compound acts as a crucial urea intermediate in the synthesis of potent antibacterial agents that overcome bacterial resistance mechanisms. The disclosed methodology represents a significant leap forward in process chemistry, replacing hazardous reagents with a safer, silicon-mediated approach that enhances both selectivity and overall yield. By leveraging temporary silyl protection strategies combined with carbonyldiimidazole (CDI) activation, this invention addresses long-standing challenges in forming the strained 1,6-diazabicyclo[3.2.1]octane core structure efficiently.
For R&D directors and process chemists, the implications of this technology are profound, offering a route that minimizes impurity profiles while maximizing throughput. The ability to generate high-purity intermediates reliably is essential for maintaining the stringent quality standards required in modern API manufacturing. This report analyzes the technical merits of this silicon-based cyclization strategy, highlighting its potential to redefine cost structures and supply chain reliability for manufacturers of next-generation antibiotics. The transition from traditional phosgene equivalents to this greener alternative underscores a commitment to sustainable chemistry without compromising on performance metrics.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the formation of cyclic urea moieties within complex heterocyclic frameworks like the 1,6-diazabicyclo[3.2.1]octane system has relied heavily on the use of triphosgene or similar phosgene equivalents. While effective in some contexts, these reagents pose severe safety hazards due to their high toxicity and the potential release of phosgene gas during handling. Furthermore, direct carbonylation of polyfunctional diamines often suffers from poor regioselectivity, leading to a mixture of isomers and oligomeric byproducts that are difficult to separate. In the specific context of beta-lactamase inhibitor precursors, competing reactions at unprotected nitrogen atoms can drastically reduce the yield of the desired bicyclic product. These inefficiencies necessitate extensive purification steps, increasing solvent consumption, waste generation, and overall production costs, thereby creating bottlenecks in the supply chain for critical antibiotic intermediates.
The Novel Approach
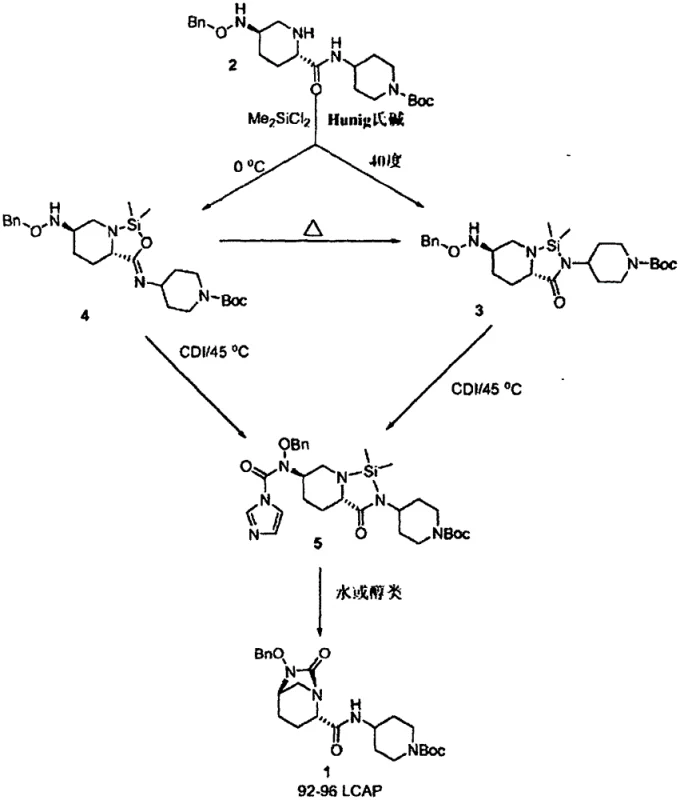
The innovative process described in the patent circumvents these issues by introducing a silicon-containing compound, such as dichlorodimethylsilane, prior to the carbonylation step. This strategic modification allows for the in situ formation of a five-membered N-Si-N or N-Si-O intermediate, which effectively masks reactive sites and directs the subsequent reaction with CDI exclusively towards the desired urea formation. This temporary protection strategy ensures that the carbonylation occurs with high fidelity, cleanly converting the intermediate into the target urea structure upon hydrolysis. The result is a process that not only eliminates the need for hazardous triphosgene but also delivers the final urea intermediate in approximately 90% yield with exceptional purity. This shift from a hazard-intensive protocol to a controlled, selective synthesis demonstrates a mature understanding of protective group chemistry applied to industrial scale-up.
Mechanistic Insights into Silicon-Mediated Cyclization
The core of this technological advancement lies in the precise manipulation of nucleophilicity through silylation. When the diamine precursor reacts with a dihalodialkylsilane in the presence of a base like Hunig's base, it forms a transient silylated species. This species is pivotal because it alters the electronic environment of the nitrogen atoms, preventing unwanted side reactions during the subsequent addition of the carbonyl source. The use of CDI as the carbonylating agent is particularly advantageous here; unlike triphosgene, CDI is a solid, easier-to-handle reagent that releases imidazole as a byproduct, which is less corrosive and easier to remove. The reaction proceeds through a well-defined pathway where the silyl group stabilizes the transition state, ensuring that the carbonyl group is inserted exactly where needed to close the ring upon subsequent hydrolysis.
Furthermore, the hydrolysis step itself is elegantly designed to trigger spontaneous cyclization. By treating the carbonylated silyl-intermediate with a protic solvent like isopropanol or water, the Si-N bonds are cleaved. This deprotection unmasks the reactive amine functionality, which immediately attacks the adjacent carbonyl carbon to form the stable bicyclic urea ring. This cascade effect minimizes the residence time of reactive intermediates, thereby reducing the opportunity for degradation or epimerization. For quality control teams, this mechanism translates to a cleaner reaction profile with fewer unknown impurities, simplifying the analytical burden and ensuring that the material meets the rigorous specifications required for downstream coupling with beta-lactam antibiotics.
How to Synthesize Tert-butyl 4-((2S,5R)-6-(benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboxamido)piperidine-1-carboxylate Efficiently
The synthesis protocol outlined in the patent provides a clear, three-step sequence that is amenable to large-scale manufacturing. It begins with the silylation of the diamine starting material in acetonitrile at controlled low temperatures to ensure selectivity. This is followed by the addition of CDI at moderately elevated temperatures to drive the carbonylation to completion. Finally, the reaction mixture is treated with isopropanol to hydrolyze the silyl groups and induce cyclization, yielding the final product after a straightforward workup involving extraction and crystallization. This streamlined approach reduces the number of unit operations compared to traditional methods, directly impacting the operational efficiency of the production line.
- React the diamine precursor with dichlorodimethylsilane and Hunig's base in acetonitrile at controlled low temperatures to form silylated intermediates.
- Treat the silylated mixture with carbonyldiimidazole (CDI) at elevated temperatures (approx. 45°C) to effect carbonylation and form the protected urea species.
- Hydrolyze the silyl groups using isopropanol or water to trigger spontaneous cyclization, yielding the final bicyclic urea product with high purity.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this silicon-mediated process offers substantial benefits that extend beyond mere chemical elegance. For procurement managers, the replacement of triphosgene with dichlorodimethylsilane and CDI represents a significant reduction in regulatory burden and safety compliance costs. Triphosgene is a scheduled substance in many jurisdictions, requiring specialized storage, handling protocols, and emergency response plans. By eliminating this hazard, manufacturers can lower their insurance premiums and reduce the capital expenditure associated with safety infrastructure. Moreover, the reagents used in this new process are commodity chemicals with stable supply chains, mitigating the risk of raw material shortages that could disrupt production schedules.
- Cost Reduction in Manufacturing: The improved yield of approximately 90% for the urea intermediate directly translates to lower cost of goods sold (COGS). Higher yields mean less starting material is wasted, and the reduced formation of byproducts minimizes the need for expensive chromatographic purification steps. Additionally, the use of simpler workup procedures, such as phase cuts and crystallization rather than complex distillations or column chromatography, reduces solvent consumption and energy usage. These cumulative efficiencies result in a more cost-effective manufacturing process, allowing for competitive pricing in the global market for antibiotic intermediates without sacrificing margin.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions, which tolerate a range of temperatures and reagent ratios, ensures consistent batch-to-batch quality. This reliability is critical for supply chain heads who must guarantee uninterrupted delivery to API manufacturers. The process avoids sensitive catalysts that might require inert atmospheres or ultra-dry conditions, making it easier to execute in standard multipurpose reactors. Consequently, lead times can be shortened, and inventory planning becomes more predictable, fostering stronger partnerships between intermediate suppliers and pharmaceutical clients.
- Scalability and Environmental Compliance: Scaling chemical processes often amplifies safety and environmental risks, but this methodology is inherently safer and greener. The absence of phosgene gas generation and the use of less toxic reagents align with modern green chemistry principles, facilitating easier permitting and environmental compliance. The waste stream is less hazardous, reducing disposal costs and environmental impact. This sustainability profile is increasingly important for pharmaceutical companies aiming to meet their corporate social responsibility goals, making suppliers who utilize this technology preferred partners for long-term contracts.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthesis route. Understanding these details helps stakeholders evaluate the feasibility of integrating this intermediate into their existing supply chains. The answers are derived directly from the experimental data and embodiments provided in the patent documentation, ensuring accuracy and relevance for technical decision-makers.
Q: Why is the silicon-mediated method superior to triphosgene for this synthesis?
A: The silicon-mediated method avoids the use of highly toxic triphosgene, significantly improving process safety. Additionally, the temporary silyl protection ensures high regioselectivity during carbonylation, preventing side reactions common with direct CDI treatment and resulting in yields around 90%.
Q: What are the critical reaction conditions for the silylation step?
A: The silylation step requires strict temperature control, typically between -10°C and 20°C, using a base like Hunig's base in acetonitrile. This ensures the formation of the correct N-Si-N or N-Si-O intermediates without degradation.
Q: How does this process impact downstream purification?
A: The process yields a crude product with high purity (>98 LCAP) after simple workup. The material demonstrates excellent performance in subsequent steps, such as palladium-catalyzed debenzylation, ensuring consistent quality for the final API.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tert-butyl 4-((2S,5R)-6-(benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboxamido)piperidine-1-carboxylate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the development of life-saving antibiotics. Our technical team has extensively evaluated the silicon-mediated pathway described in CN107428784B and confirmed its viability for commercial production. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the volumetric demands of global pharmaceutical partners. Our facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, including the >98 LCAP purity demonstrated in the patent examples, guaranteeing that every batch meets the exacting standards required for GMP API synthesis.
We invite procurement leaders and R&D directors to engage with us for a Customized Cost-Saving Analysis tailored to your specific project needs. By leveraging this advanced synthetic route, we can help you optimize your supply chain for both cost and reliability. Please contact our technical procurement team to request specific COA data and route feasibility assessments. We are committed to being your strategic partner in navigating the complexities of antibiotic intermediate manufacturing, delivering value through innovation and operational excellence.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →