Scalable Synthesis of 2,6-Difluoro-3,5-Dimethoxybenzyl Alcohol for Global Pharma Supply Chains
Scalable Synthesis of 2,6-Difluoro-3,5-Dimethoxybenzyl Alcohol for Global Pharma Supply Chains
The rapid advancement of targeted cancer therapies has placed Fibroblast Growth Factor Receptor (FGFR) inhibitors at the forefront of oncology research, creating an urgent demand for high-quality key intermediates. Patent CN115368219A discloses a groundbreaking preparation method for 2,6-difluoro-3,5-dimethoxybenzyl alcohol, a critical building block for FGFR inhibitors such as the clinical candidate ASP-5878. This novel synthetic route addresses long-standing challenges in process chemistry by utilizing a strategic decarboxylation approach rather than direct fluorination, thereby eliminating complex isomer separation steps. For pharmaceutical manufacturers, this represents a significant leap forward in process efficiency, offering a pathway to secure, high-purity supply chains for next-generation antitumor drugs. The methodology transforms a traditionally cumbersome synthesis into a streamlined, industrially viable operation that aligns perfectly with modern green chemistry principles.
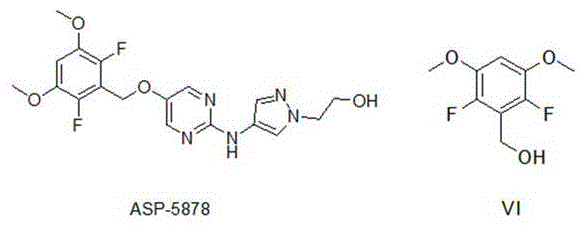
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of fluorinated aromatic intermediates has relied heavily on direct electrophilic or nucleophilic fluorination of pre-functionalized benzene rings, a strategy fraught with significant technical hurdles. As illustrated in prior art reports, attempting to introduce fluorine atoms onto a dimethoxy-substituted benzene scaffold often results in poor regioselectivity, generating a mixture of mono-fluorinated and poly-fluorinated isomers that are notoriously difficult to separate. This lack of selectivity not only drives down the overall yield but also necessitates extensive and costly purification processes, such as preparative HPLC or repeated recrystallization, which are impractical for multi-kilogram production. Furthermore, the use of aggressive fluorinating reagents poses safety risks and generates substantial hazardous waste, complicating environmental compliance and increasing the total cost of ownership for the manufacturing process.
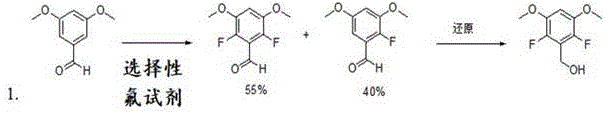
The Novel Approach
In stark contrast, the method disclosed in CN115368219A adopts a clever retrosynthetic strategy that builds the substitution pattern through nucleophilic aromatic substitution and decarboxylation, effectively bypassing the selectivity issues of direct fluorination. By starting with commercially available 2,3,5,6-tetrafluorobenzoic acid, the process leverages the inherent activation of the ring to selectively replace specific fluorine atoms with methoxy groups under controlled conditions. The subsequent removal of the carboxyl group via decarboxylation serves as a powerful driving force that simplifies the molecular architecture without disturbing the carefully installed substitution pattern. This approach ensures that the final product is obtained with high regiochemical purity, eliminating the need for difficult isomer separations and drastically reducing the number of unit operations required. The result is a robust, linear synthesis that is inherently safer, cleaner, and more economically attractive for large-scale industrial application.
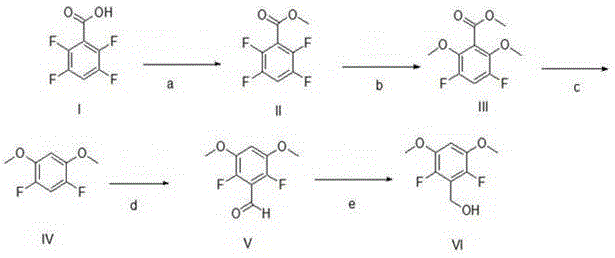
Mechanistic Insights into the Decarboxylation and Lithiation Strategy
The core of this innovative synthesis lies in the precise control of nucleophilic aromatic substitution (SnAr) and the subsequent thermal decarboxylation, which together dictate the success of the entire pathway. In the second step, sodium methoxide acts as a potent nucleophile, attacking the electron-deficient aromatic ring at the positions ortho and para to the ester group, displacing fluorine atoms to install the requisite methoxy groups. This reaction is highly dependent on temperature control, typically maintained between 50°C and 80°C, to ensure complete conversion while preventing side reactions. Following this, the decarboxylation step utilizes potassium fluoride or cesium fluoride in polar aprotic solvents like DMSO at elevated temperatures (80°C to 120°C). The fluoride ion likely facilitates the loss of carbon dioxide by stabilizing the transition state or acting as a base, resulting in the formation of the 1,3-difluoro-2,4-dimethoxybenzene core with exceptional fidelity.
The final functionalization of the aromatic core involves a sophisticated sequence of lithiation and reduction that requires rigorous exclusion of moisture and oxygen to succeed. The introduction of the hydroxymethyl group begins with the deprotonation of the aromatic ring using lithium diisopropylamide (LDA) at cryogenic temperatures, specifically around -78°C, to generate a stable aryl lithium species. This highly reactive intermediate is then quenched with N,N-dimethylformamide (DMF), effectively installing an aldehyde group at the desired position through a formylation mechanism. The final reduction using sodium borohydride in methanol is a mild and selective transformation that converts the aldehyde to the primary alcohol without affecting the sensitive fluorine substituents. This step-wise mechanistic progression ensures that each functional group is introduced with maximum precision, minimizing the formation of by-products and guaranteeing a consistent quality profile for the final intermediate.
How to Synthesize 2,6-Difluoro-3,5-Dimethoxybenzyl Alcohol Efficiently
The execution of this synthesis requires careful attention to reaction parameters, particularly temperature and stoichiometry, to replicate the high yields reported in the patent literature. The process begins with the esterification of the starting acid, followed by the critical methoxylation and decarboxylation steps which set the substitution pattern. Operators must ensure that the lithiation step is performed under strictly anhydrous conditions to prevent quenching of the organolithium intermediate, which would lead to significant yield loss. Detailed standard operating procedures regarding reagent addition rates, cooling profiles, and work-up protocols are essential for transferring this chemistry from the laboratory to the pilot plant. For a comprehensive breakdown of the specific experimental conditions and isolation techniques, please refer to the standardized guide below.
- Esterify 2,3,5,6-tetrafluorobenzoic acid with thionyl chloride in methanol to form methyl 2,3,5,6-tetrafluorobenzoate.
- Perform nucleophilic substitution using sodium methoxide to replace specific fluorine atoms with methoxy groups.
- Execute thermal decarboxylation using potassium fluoride in DMSO to remove the carboxyl group.
- Conduct low-temperature lithiation followed by DMF quenching to introduce the aldehyde functionality.
- Reduce the resulting aldehyde using sodium borohydride in methanol to obtain the final benzyl alcohol product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthetic route offers tangible benefits that extend far beyond simple chemical yield improvements. By eliminating the need for complex isomer separation and hazardous fluorination reagents, the process significantly reduces the consumption of raw materials and solvents, leading to a leaner and more cost-effective manufacturing model. The reliance on commodity chemicals such as sodium methoxide, methanol, and sodium borohydride ensures that the supply chain remains resilient against market volatility, as these reagents are widely available from multiple global suppliers. Furthermore, the simplified purification workflow reduces the burden on waste treatment facilities and shortens the overall production cycle time, allowing for faster turnaround on orders and improved responsiveness to customer demand fluctuations.
- Cost Reduction in Manufacturing: The elimination of expensive fluorinating agents and the reduction in purification steps directly translate to lower variable costs per kilogram of product. By avoiding the generation of hard-to-separate impurities, the process minimizes product loss during isolation, effectively increasing the mass balance and reducing the cost of goods sold. Additionally, the use of standard industrial solvents and reagents avoids the premium pricing associated with specialized fluorine chemistry, providing a structural cost advantage over legacy methods.
- Enhanced Supply Chain Reliability: Starting from 2,3,5,6-tetrafluorobenzoic acid, a stable and commercially accessible feedstock, mitigates the risk of raw material shortages that often plague specialized fluorine chemistry supply chains. The robustness of the reaction conditions means that production is less susceptible to minor variations in utility availability or equipment performance, ensuring a consistent and reliable output of high-purity intermediate. This stability is crucial for maintaining uninterrupted supply to downstream API manufacturers who operate on tight just-in-time schedules.
- Scalability and Environmental Compliance: The process is designed with scale-up in mind, utilizing reaction conditions that are easily managed in standard stainless steel reactors without requiring exotic materials of construction. The avoidance of hazardous gases and the generation of less toxic waste streams simplify environmental permitting and reduce the operational overhead associated with waste disposal. This alignment with green chemistry principles not only lowers regulatory risk but also enhances the sustainability profile of the final pharmaceutical product, a factor of increasing importance to global healthcare stakeholders.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route in an industrial setting. These answers are derived directly from the experimental data and technical specifications outlined in the patent documentation, providing a clear understanding of the process capabilities. Understanding these details is vital for technical teams evaluating the feasibility of adopting this new method for their specific production needs.
Q: What are the key advantages of this new synthesis route over traditional fluorination methods?
A: The new route avoids the difficult separation of monofluoro isomers common in direct fluorination, significantly improving purity and overall yield while reducing waste generation.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the method utilizes standard industrial reagents like sodium methoxide and sodium borohydride, operates at manageable temperatures, and avoids hazardous fluorinating gases, making it highly scalable.
Q: What is the expected purity profile of the intermediate produced via this method?
A: The process is designed to minimize impurities, particularly regioisomers, achieving high purity levels (e.g., >97% in initial steps) without complex chromatographic purification.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2,6-Difluoro-3,5-Dimethoxybenzyl Alcohol Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of FGFR inhibitors depends on the availability of high-quality intermediates produced via robust and scalable pathways. Our team of expert process chemists has thoroughly analyzed this novel decarboxylation route and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring this technology to fruition. We are committed to delivering 2,6-difluoro-3,5-dimethoxybenzyl alcohol with stringent purity specifications, supported by our rigorous QC labs that ensure every batch meets the exacting standards of the global pharmaceutical industry. Our facility is equipped to handle the specific thermal and safety requirements of the lithiation and decarboxylation steps, guaranteeing a seamless transition from development to full-scale manufacturing.
We invite procurement leaders and R&D directors to engage with us to explore how this optimized synthesis can drive value in your supply chain. By partnering with us, you gain access to a Customized Cost-Saving Analysis that quantifies the economic benefits of switching to this superior route. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments tailored to your project timelines. Let us help you secure a competitive advantage through superior chemistry and reliable supply.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →