Revolutionizing Oral Aromatase Inhibitor Production Through Advanced Steroidal Intermediate Synthesis
Revolutionizing Oral Aromatase Inhibitor Production Through Advanced Steroidal Intermediate Synthesis
The pharmaceutical landscape for hormone-dependent oncology treatments has long sought robust solutions that balance potent biological activity with practical administration routes. Patent CN86104664A presents a groundbreaking methodology for synthesizing 6-alkylidene-androsta-1,4-diene-3,17-dione compounds, specifically targeting the inhibition of aromatase enzymes responsible for estrogen biosynthesis. This technology addresses a critical gap in previous generations of inhibitors, which often suffered from poor oral bioavailability due to rapid metabolic clearance. By establishing a stable 1,4-diene conjugated system within the steroidal backbone, this invention enables the production of compounds like 6-methyleneandrost-1,4-diene-3,17-dione (FCE 24304) that maintain high therapeutic efficacy when administered orally. For R&D directors and procurement specialists, understanding the chemical nuances of this pathway is essential for securing a reliable pharmaceutical intermediates supplier capable of delivering high-purity materials for next-generation cancer therapies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art in the field of aromatase inhibition relied heavily on compounds such as 4-hydroxyandrost-4-ene-3,17-dione (4-OH-A), which demonstrated potent in vitro activity but failed to translate effectively into clinical settings via oral delivery. The fundamental chemical limitation of these earlier molecules lies in their susceptibility to hepatic metabolism, necessitating parenteral administration methods that are inconvenient for long-term patient care and increase overall treatment costs. Furthermore, conventional synthetic routes often involved complex multi-step sequences with low overall yields, utilizing expensive transition metal catalysts that introduced significant challenges in downstream purification and heavy metal residue control. These factors collectively created substantial bottlenecks in cost reduction in API manufacturing, making large-scale production economically unfeasible for many generic manufacturers seeking to enter the endocrine therapy market.
The Novel Approach
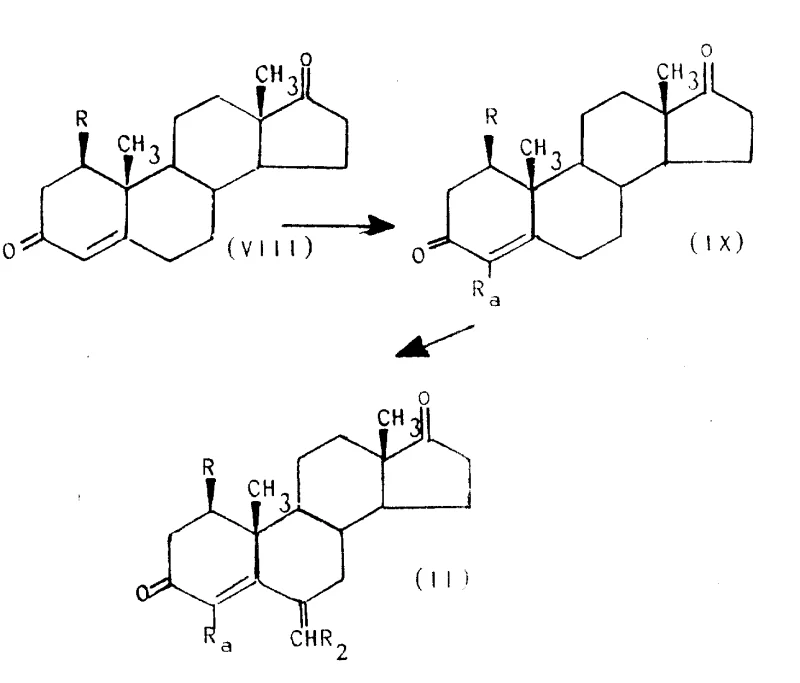
The methodology disclosed in CN86104664A overcomes these historical barriers by introducing a streamlined dehydrogenation strategy that constructs the critical 1,4-diene motif with high regioselectivity. By utilizing oxidants such as dichlorodicyanobenzoquinone (DDQ) or selenium dioxide under controlled thermal conditions, the process efficiently converts saturated or mono-unsaturated precursors into the desired conjugated system without compromising the integrity of the sensitive ketone functionalities at positions 3 and 17. This chemical innovation not only improves the metabolic stability of the final drug substance but also simplifies the synthetic workflow, thereby enhancing supply chain reliability for global pharmaceutical partners. The ability to produce these complex steroidal structures through fewer steps with higher purity directly correlates to substantial cost savings and reduced lead times for high-purity pharmaceutical intermediates.
Mechanistic Insights into DDQ-Catalyzed Dehydrogenation
The core of this technological advancement lies in the precise manipulation of the steroidal A-ring to achieve the 1,4-diene configuration. The reaction mechanism involves the abstraction of hydride ions from the C-1 and C-2 positions of the precursor Formula (II) compound, facilitated by the electron-deficient nature of the DDQ reagent. This dehydrogenation proceeds through a concerted pathway that preserves the stereochemistry at the C-10 methyl group while establishing the necessary conjugation for aromatase binding affinity. The choice of solvent plays a pivotal role in this transformation; inert solvents such as dioxane, benzene, or toluene provide the optimal dielectric environment to stabilize the transition state, ensuring reaction temperatures between 40°C and 120°C drive the conversion to completion without inducing unwanted side reactions or decomposition of the sensitive enone system.
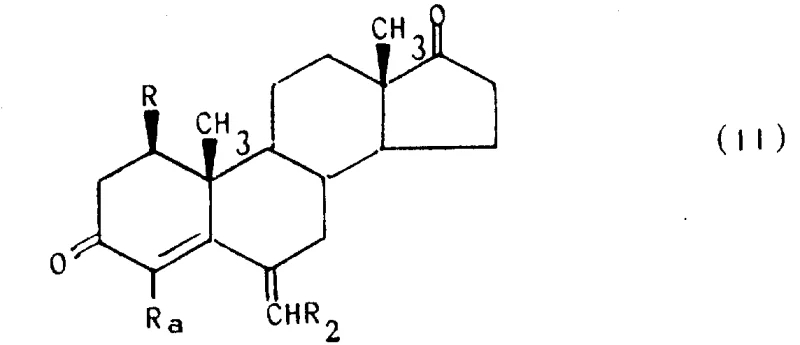
Following the initial dehydrogenation, the process allows for further functionalization through epoxidation and subsequent ring-opening reactions to introduce halogen substituents at the C-4 position. This versatility is crucial for generating a library of analogs with varying lipophilicity and receptor binding profiles. The epoxidation step utilizes alkaline hydrogen peroxide to selectively target the C-4,5 double bond, creating an oxirane intermediate that serves as a versatile handle for nucleophilic attack. Subsequent treatment with hydrogen halides or boron trihalides opens the epoxide ring with high stereoselectivity, yielding 4-halo derivatives that exhibit enhanced potency. This modular approach to structural modification ensures that the commercial scale-up of complex hormonal agents remains flexible, allowing manufacturers to adapt quickly to evolving clinical requirements without retooling entire production lines.
How to Synthesize 6-Methyleneandrost-1,4-diene-3,17-dione Efficiently
Executing this synthesis requires strict adherence to the reaction parameters outlined in the patent to ensure consistent quality and yield. The process begins with the preparation of the 6-alkylidene precursor, which involves the condensation of a 3-keto steroid with formaldehyde acetals in the presence of acidic catalysts such as phosphorus oxychloride. This step must be carefully monitored to prevent polymerization of the formaldehyde source and to ensure the exclusive formation of the exocyclic methylene group at the C-6 position. Once the precursor is secured, the dehydrogenation reaction is initiated by adding DDQ in a molar excess to drive the equilibrium towards the product, followed by rigorous workup procedures involving alumina filtration to remove reduced quinone byproducts. Detailed standardized synthesis steps are provided below to guide process chemists in replicating these results at scale.
- Prepare the precursor Formula (II) compound through alkylation of androst-4-ene-3,17-dione followed by condensation with formaldehyde acetals.
- Execute dehydrogenation using DDQ in refluxing dioxane or selenium dioxide in tert-butanol to establish the 1,4-diene system.
- Purify the final crude product via silica gel chromatography using hexane and ethyl acetate gradients to isolate high-purity isomers.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic sourcing perspective, the adoption of this synthetic route offers profound benefits for procurement managers focused on optimizing the cost structure of oncology API production. The elimination of expensive transition metal catalysts, which are often required in alternative cross-coupling strategies for similar structures, removes the need for costly scavenging resins and extensive analytical testing for residual metals. This simplification of the purification train translates directly into cost reduction in API manufacturing, as it reduces both material consumption and processing time. Furthermore, the reagents utilized in this protocol, such as DDQ and common organic solvents, are readily available from multiple global suppliers, mitigating the risk of single-source dependency and ensuring enhanced supply chain reliability even during periods of market volatility.
- Cost Reduction in Manufacturing: The streamlined nature of the dehydrogenation and halogenation sequence significantly reduces the number of unit operations required to reach the final intermediate. By avoiding complex protection and deprotection strategies often seen in steroidal chemistry, the process minimizes solvent usage and waste generation, leading to substantial cost savings in raw material procurement and waste disposal. The high selectivity of the reaction also reduces the burden on chromatographic purification, allowing for more efficient use of silica gel and eluents, which are significant cost drivers in large-scale pharmaceutical production.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions, which tolerate a reasonable range of temperatures and stoichiometry, makes this process highly suitable for transfer between different manufacturing sites. This flexibility ensures reducing lead time for high-purity pharmaceutical intermediates, as production can be scaled or shifted without extensive re-validation efforts. The use of stable starting materials that do not require cryogenic storage further simplifies logistics, allowing for just-in-time inventory management and reducing the capital tied up in raw material stockpiles.
- Scalability and Environmental Compliance: The synthetic pathway is designed with scalability in mind, utilizing reaction vessels and equipment standard in the fine chemical industry. The absence of highly toxic heavy metals simplifies environmental compliance and wastewater treatment, aligning with modern green chemistry principles. This environmental advantage not only reduces regulatory risk but also enhances the corporate sustainability profile of the manufacturing partner, a key consideration for multinational pharmaceutical companies committed to responsible sourcing practices.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented technology. These insights are derived directly from the experimental data and claims within CN86104664A, providing a factual basis for decision-making. Understanding these details is vital for technical teams evaluating the feasibility of integrating this intermediate into their existing drug development pipelines.
Q: Why is the 1,4-diene structure critical for oral bioavailability in this patent?
A: The introduction of the 1,4-diene system significantly enhances metabolic stability compared to prior art 4-ene compounds, preventing rapid hepatic degradation and allowing effective oral administration.
Q: What are the preferred reagents for the dehydrogenation step?
A: The patent identifies dichlorodicyanobenzoquinone (DDQ) in inert solvents like dioxane or toluene as the optimal system, though selenium dioxide and chloranil are also viable alternatives depending on scale.
Q: How does this synthesis address impurity control?
A: The process utilizes specific crystallization and chromatography techniques to separate Z and E isomers, ensuring the final API intermediate meets stringent purity specifications required for oncology applications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 6-Methyleneandrost-1,4-diene-3,17-dione Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of securing a supply chain for complex steroidal intermediates that meets the rigorous demands of modern oncology drug development. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition seamlessly from clinical trials to full-scale market launch. We maintain stringent purity specifications and operate rigorous QC labs equipped with advanced analytical instrumentation to verify the identity and quality of every batch, guaranteeing that the 6-methyleneandrost-1,4-diene-3,17-dione supplied meets all regulatory requirements for API synthesis.
We invite you to engage with our technical procurement team to discuss how this innovative synthetic route can optimize your specific manufacturing needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the potential economic benefits of switching to this more efficient pathway. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your project timeline, ensuring that your supply of high-purity steroid intermediates remains uninterrupted and cost-effective.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →