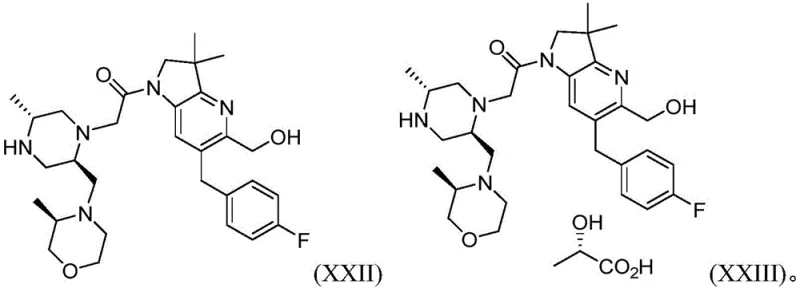
Advanced Synthesis of IAP Antagonist Intermediate XXIII for Commercial Pharmaceutical Manufacturing
The pharmaceutical industry continuously seeks robust synthetic routes for complex oncology targets, and patent CN115968370A presents a significant advancement in the manufacturing of IAP antagonist compounds. This intellectual property details an improved process for preparing compounds of Formula XXIII and its intermediates, specifically addressing the limitations of previous synthetic methodologies that relied on unstable reagents and expensive catalysts. The core innovation lies in the strategic use of palladium-catalyzed carbonylation to generate key crystalline intermediates, which fundamentally alters the impurity profile and physical properties of the final active pharmaceutical ingredient. By shifting away from pyrophoric reagents and unpredictable coupling conditions, this technology offers a pathway to higher purity standards essential for clinical and commercial success. The structural complexity of the IAP antagonist requires precise stereochemical control, which is maintained throughout the novel sequence described in the patent documentation. This report analyzes the technical merits of this synthesis from the perspective of process chemistry and commercial viability for global supply chains.
The limitations of conventional methods for synthesizing IAP antagonists often stem from the reliance on reagents that are difficult to handle on a large scale, such as tert-butyl lithium or specialized zinc species like compound Formula VI. These materials are not only hazardous due to their flammability and pyrophoricity but also suffer from limited commercial availability and batch-to-batch variability, which introduces significant risk into the manufacturing timeline. Furthermore, prior art routes, such as those disclosed in U.S. Patent No. 9,783,538, frequently result in lower yields and the formation of persistent impurities, specifically bis-hydroxymethyl byproducts that are challenging to purge during downstream processing. The use of PEPPSI catalysts in earlier iterations also contributed to high production costs and complicated metal removal protocols, creating bottlenecks for procurement teams aiming to reduce the cost of goods sold. These technical debt issues in legacy processes necessitate a re-evaluation of the synthetic strategy to ensure supply chain resilience and regulatory compliance.
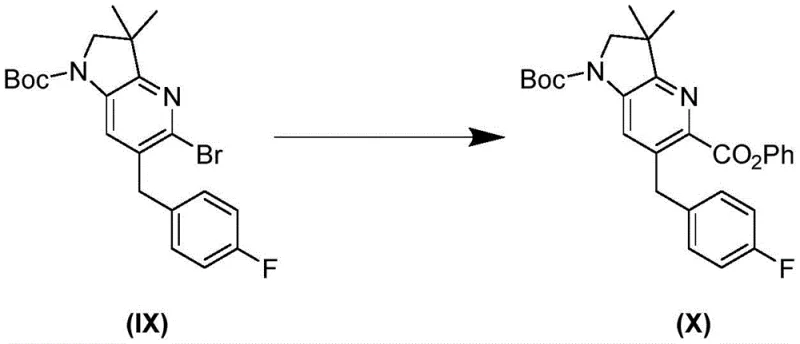
The novel approach disclosed in CN115968370A overcomes these hurdles by introducing a streamlined sequence that utilizes stable, commercially available starting materials and avoids the problematic zinc reagents entirely. A pivotal improvement is the conversion of compound Formula IX to compound Formula X via palladium-catalyzed carbonylation, which generates a highly crystalline phenyl ester intermediate. This crystallinity is a critical process attribute, as it allows for efficient purification through crystallization rather than relying solely on chromatography, which is often impractical at multi-kilogram scales. The new route also incorporates a convergent coupling strategy between Formula XIII and Formula XX, which maximizes atom economy and simplifies the overall process flow. By eliminating the need for hazardous reagents and improving the physical form of intermediates, this methodology significantly enhances the safety profile and operational efficiency of the manufacturing plant. The result is a more predictable synthesis that delivers the final L-(+)-lactate salt of Formula XXIII with superior purity and stability characteristics.
Mechanistic Insights into Palladium-Catalyzed Carbonylation and Coupling
The mechanistic foundation of this improved synthesis rests on the efficient transformation of the brominated intermediate Formula IX into the ester Formula X using carbon monoxide and a palladium catalyst system. This carbonylation step is meticulously optimized to proceed under mild conditions, typically utilizing ligands such as rac-BINAP or XPhos to facilitate the oxidative addition and migratory insertion steps required for ester formation. The choice of phenol or phenyl formate as the carbonyl source allows for the generation of a stable ester that serves as a protected form of the hydroxymethyl group, preventing premature oxidation or side reactions. This mechanistic choice is crucial because it avoids the direct handling of unstable aldehyde intermediates that often lead to the formation of the problematic Formula XXV impurity observed in storage stability studies. The reaction conditions are tuned to ensure complete conversion while minimizing the formation of dehalogenated byproducts, thereby maintaining a high mass balance throughout the sequence. This level of control over the reaction pathway is essential for meeting the stringent impurity specifications required for oncology drug substances.
Impurity control is further enhanced in the subsequent coupling and deprotection stages, where the process design actively mitigates the risk of racemization and degradation. The coupling of the chloroacetyl derivative Formula XIII with the piperazine intermediate Formula XX is conducted under basic conditions that favor nucleophilic substitution without compromising the stereochemical integrity of the chiral centers. Following the coupling, the deprotection of the Boc group is managed using acidic conditions that are carefully quenched to prevent acid-catalyzed decomposition of the sensitive pyrrolo-pyridine core. The final salt formation with anhydrous L-lactic acid is performed in a specific solvent system, often involving ethyl acetate and heptane, to induce the crystallization of the thermodynamically stable Form C. This specific polymorph is critical for long-term stability, as it demonstrates significantly lower levels of aldehyde impurity generation over time compared to amorphous forms. The entire mechanistic sequence is designed to minimize the presence of residual palladium, which has been correlated with oxidative degradation, ensuring that the final drug substance remains stable under standard storage conditions of 25 degrees Celsius and 60 percent relative humidity.
How to Synthesize IAP Antagonist Intermediate Efficiently
The synthesis of the target IAP antagonist intermediate requires a disciplined approach to reaction conditions and purification protocols to ensure the high purity and yield described in the patent literature. The process begins with the preparation of the key brominated building block, followed by the critical carbonylation step that defines the efficiency of the entire route. Operators must pay close attention to the control of carbon monoxide pressure and temperature during the ester formation to maximize the crystallinity of the intermediate. Subsequent steps involve the precise stoichiometric addition of coupling partners and the careful management of exotherms during the salt formation phase. Detailed standard operating procedures for each transformation are essential to replicate the success of the laboratory scale in a commercial manufacturing environment. The following guide outlines the standardized synthesis steps derived from the exemplary embodiments.
- Preparation of key intermediate Formula IX via bromination and protection strategies avoiding unstable zinc reagents.
- Palladium-catalyzed carbonylation of Formula IX to generate crystalline ester intermediate Formula X.
- Convergent coupling of Formula XIII and Formula XX followed by deprotection and L-lactic acid salt formation.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this novel synthetic route offers substantial benefits for procurement managers and supply chain directors focused on cost reduction and reliability. The elimination of expensive and hazardous reagents like PEPPSI catalysts and compound Formula VI directly translates to a lower raw material cost base, as the new process relies on commodity chemicals that are readily available from multiple global suppliers. This diversification of the supply base reduces the risk of single-source bottlenecks and provides greater leverage in vendor negotiations. Furthermore, the improved yield and purity profiles mean that less material is wasted during purification, effectively increasing the overall throughput of the manufacturing facility without the need for capital expansion. The ability to produce a crystalline intermediate also simplifies logistics, as solids are generally easier and safer to transport and store than unstable oils or solutions. These factors combine to create a more resilient supply chain capable of meeting the demanding schedules of clinical trials and commercial launch.
- Cost Reduction in Manufacturing: The process achieves significant cost savings by replacing proprietary and high-cost catalysts with standard palladium systems and avoiding the use of cryogenic reagents that require specialized handling infrastructure. The improved efficiency of the carbonylation step reduces the consumption of starting materials, while the enhanced purity of intermediates minimizes the need for extensive chromatographic purification, which is a major cost driver in fine chemical manufacturing. Additionally, the higher overall yield of the sequence means that more final product is obtained per batch, effectively lowering the cost per kilogram of the active pharmaceutical ingredient. These qualitative improvements in process efficiency contribute to a more competitive pricing structure for the final drug product.
- Enhanced Supply Chain Reliability: By utilizing stable intermediates and avoiding reagents with short shelf lives, the manufacturing process becomes less susceptible to disruptions caused by material degradation or supply shortages. The robustness of the synthetic route allows for longer campaign runs and reduces the frequency of batch failures, ensuring a consistent flow of material to downstream formulation teams. The use of common solvents and reagents also simplifies the procurement process, as these items can be sourced from established chemical distributors with reliable delivery track records. This stability is crucial for maintaining the continuity of supply required for long-term commercial contracts and regulatory filings.
- Scalability and Environmental Compliance: The novel method is designed with scale-up in mind, featuring unit operations such as crystallization and filtration that are easily transferred from pilot plant to commercial production scales. The reduction in hazardous waste generation, particularly from the avoidance of pyrophoric reagents, aligns with increasingly stringent environmental regulations and corporate sustainability goals. The process also facilitates easier waste stream management, as the solvent systems used are amenable to recovery and recycling. This environmental compatibility not only reduces disposal costs but also enhances the company's reputation as a responsible manufacturer, which is an important factor for partnerships with major pharmaceutical companies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and supply of IAP antagonist intermediates, based on the data provided in the patent documentation. These answers are derived from the specific experimental examples and comparative data presented in the intellectual property, offering clarity on the advantages of the new method. Understanding these details is vital for technical teams evaluating the feasibility of this route for their specific development programs. The information provided here serves as a preliminary guide for further discussion with our technical procurement team.
Q: How does the new synthesis route improve impurity control compared to prior art?
A: The disclosed method avoids the use of PEPPSI catalysts and unstable compound Formula VI, significantly reducing bis-hydroxymethyl impurities and aldehyde degradation products like Formula XXV.
Q: What are the scalability advantages of the palladium-catalyzed carbonylation step?
A: The carbonylation step produces a highly crystalline ester intermediate Formula X, which facilitates easier isolation, purification, and handling during commercial scale-up compared to amorphous oils.
Q: How is palladium content managed in the final API to ensure stability?
A: The process includes specific purification steps and crystallization conditions that reduce palladium content to less than 10ppm, correlating with lower oxidative degradation and improved shelf-life stability.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable IAP Antagonist Intermediate Supplier
NINGBO INNO PHARMCHEM stands ready to support your development programs with our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our technical team possesses the expertise to implement the advanced carbonylation and coupling strategies described in CN115968370A, ensuring that your project benefits from the highest standards of purity and efficiency. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of IAP antagonist intermediate meets the exacting requirements of global regulatory agencies. Our commitment to quality and reliability makes us the ideal partner for navigating the complexities of oncology drug manufacturing.
We invite you to contact our technical procurement team to request a Customized Cost-Saving Analysis for your specific project needs. By collaborating with us, you can gain access to specific COA data and route feasibility assessments that will help you optimize your supply chain and reduce time to market. Let us demonstrate how our manufacturing capabilities can enhance the commercial viability of your IAP antagonist program.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →