Advanced Rh(III) Catalytic Strategy for Efficient Nitrogen Heterocycle Manufacturing
Advanced Rh(III) Catalytic Strategy for Efficient Nitrogen Heterocycle Manufacturing
The landscape of pharmaceutical intermediate synthesis is constantly evolving, driven by the need for more efficient, atom-economical, and scalable processes. A significant breakthrough in this domain is documented in patent CN109232529B, which details a novel preparation method for constructing nitrogen heterocyclic skeletons, specifically isoindolinones, using a self-assembled directing group-assisted Rh(III) catalytic system. This technology represents a paradigm shift from traditional multi-step functionalization to a streamlined one-pot protocol. By leveraging the unique ability of rhodium catalysts to activate C-H bonds in the presence of an in-situ generated imine directing group, this method allows for the direct coupling of simple aromatic aldehydes, 2-aminopyridine, and substituted olefins. For R&D directors and procurement managers seeking a reliable pharmaceutical intermediate supplier, understanding the mechanistic elegance and commercial viability of this route is critical for optimizing supply chains and reducing the cost of goods sold in API manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of isoindolinone scaffolds has relied heavily on pre-functionalized substrates that often require tedious synthetic preparation. Early methodologies, such as those reported by Li Xingwei's group in 2010, utilized N-phenyl-substituted arylformamides as substrates. While effective in some contexts, these approaches suffered from significant limitations, particularly when dealing with heterocyclic aromatic rings like furan or indole, where the reaction often stalled at the oxidation Heck product stage rather than proceeding to the desired cyclization. Furthermore, subsequent improvements by Yu Jinquan's group in 2014, which employed N-perfluoro-substituted phenyl arylcarboxamides, although successful in broadening substrate scope, introduced new complexities. The requirement for specialized, electron-deficient amide substrates meant that raw materials were more expensive and less readily available, creating bottlenecks in the supply chain. These conventional routes often demanded harsh reaction conditions and exhibited lower atom economy due to the necessity of installing and subsequently removing auxiliary directing groups, thereby increasing waste generation and processing time.
The Novel Approach
In stark contrast, the methodology disclosed in CN109232529B offers a transformative solution by utilizing a self-assembly strategy that generates the directing group in situ. This approach bypasses the need for isolating unstable or difficult-to-synthesize amide intermediates. Instead, it starts from ubiquitous and inexpensive building blocks: aromatic aldehydes and 2-aminopyridine. Under the influence of a rhodium catalyst and copper acetate oxidant, these components condense to form an active imine species that immediately directs the C-H activation process. This one-pot transformation proceeds under remarkably mild conditions, typically at 80°C in acetonitrile, achieving high yields up to 90% as demonstrated in specific embodiments. The simplicity of this operation not only reduces the number of unit operations but also significantly enhances the overall process safety and environmental profile. For manufacturers focused on cost reduction in API manufacturing, this elimination of intermediate isolation steps translates directly into substantial operational savings and a more robust production workflow.
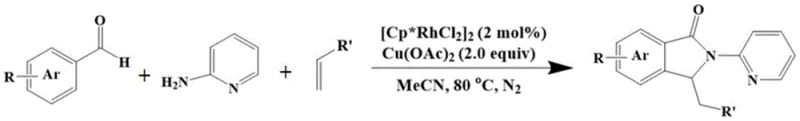
Mechanistic Insights into Rh(III)-Catalyzed C-H Activation and Cyclization
The core innovation of this technology lies in its sophisticated yet efficient catalytic cycle, which orchestrates multiple bond-forming events in a single vessel. The mechanism initiates with the condensation of the aromatic aldehyde and 2-aminopyridine to generate a Schiff base, which serves as a potent bidentate directing group. This transient species coordinates with the Rh(III) center, facilitating the crucial C-H activation step at the ortho-position of the aromatic ring. This activation creates a stable rhodacycle intermediate, setting the stage for the subsequent insertion of the substituted olefin. The electron-withdrawing nature of the substituents on the olefin, such as ester or cyano groups, plays a pivotal role in enhancing the reactivity of the double bond towards migratory insertion. Following the insertion, a beta-hydride elimination occurs, generating a Heck-type intermediate that remains coordinated to the metal center. Finally, an intramolecular Michael addition or nucleophilic attack by the nitrogen atom onto the carbonyl carbon closes the five-membered lactam ring, releasing the final isoindolinone product and regenerating the active Rh(III) catalyst for the next turnover.
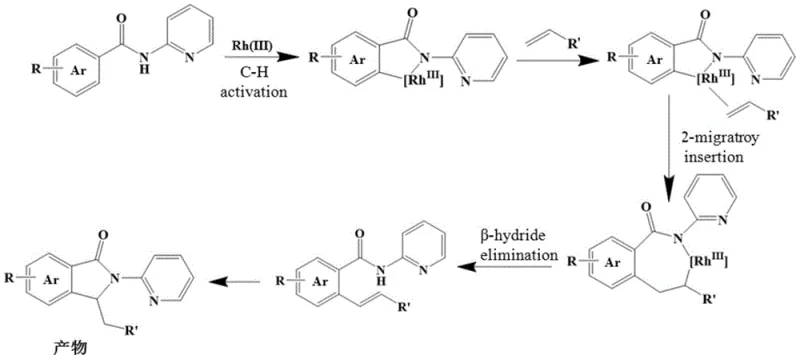
From an impurity control perspective, this mechanism offers distinct advantages over traditional radical-based or high-temperature thermal cyclizations. The directed nature of the C-H activation ensures high regioselectivity, minimizing the formation of positional isomers that are notoriously difficult to separate. Furthermore, the mild reaction temperature of 80°C prevents the thermal degradation of sensitive functional groups and suppresses non-selective polymerization of the olefin substrate. The use of copper acetate as a stoichiometric oxidant is carefully balanced to re-oxidize the Rh(I) species back to Rh(III) without causing excessive oxidative damage to the organic framework. This precise control over the redox environment results in a cleaner crude reaction profile, which simplifies downstream purification processes such as column chromatography or crystallization. For quality assurance teams, this means a more consistent impurity profile and higher confidence in meeting stringent purity specifications required for pharmaceutical applications.
How to Synthesize Isoindolinone Derivatives Efficiently
Implementing this synthesis route requires careful attention to the order of addition and atmospheric control to maximize the efficiency of the in-situ directing group formation. The process begins by charging a reaction vessel, preferably a Schlenk bottle, with the aromatic aldehyde, 2-aminopyridine, copper acetate, and the rhodium dimer catalyst. It is critical to perform vacuum-nitrogen exchange cycles to remove oxygen, which could interfere with the catalyst or oxidize sensitive intermediates prematurely. Once the inert atmosphere is established, the solvent, typically acetonitrile, is added along with the substituted olefin. The mixture is then heated to 80°C and stirred for a duration ranging from 4 to 12 hours, with reaction progress monitored via TLC. Upon completion, the solvent is removed under reduced pressure, and the residue is purified to isolate the high-purity nitrogen heterocyclic skeleton. For detailed standard operating procedures and specific molar ratios, please refer to the guide below.
- Mix aromatic aldehyde, 2-aminopyridine, copper acetate, and [Cp*RhCl2]2 in a Schlenk bottle under inert atmosphere.
- Add solvent (acetonitrile) and substituted olefin, then stir at 80°C for 4-12 hours.
- Monitor reaction by TLC, remove solvent, and purify the crude solid via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this Rh(III) catalytic technology offers compelling strategic advantages that extend beyond mere chemical novelty. The primary benefit stems from the drastic simplification of the raw material portfolio. By shifting from complex, pre-synthesized amides to commodity chemicals like benzaldehydes and aminopyridines, companies can leverage existing bulk supply contracts and reduce exposure to volatile pricing of specialty intermediates. This transition effectively de-risks the supply chain, ensuring greater continuity of supply even during market fluctuations. Moreover, the one-pot nature of the reaction eliminates the need for intermediate isolation, drying, and quality control testing between steps, which significantly reduces the overall manufacturing lead time. This streamlined workflow allows for faster batch turnover and improved asset utilization in multipurpose production facilities.
- Cost Reduction in Manufacturing: The economic impact of this process is driven by the elimination of expensive protecting group chemistry and the reduction of solvent usage associated with intermediate workups. Since the directing group is formed in situ from cheap starting materials, the cost of goods sold is significantly lowered compared to routes requiring custom-synthesized substrates. Additionally, the high atom economy of the reaction means that a larger proportion of the input mass ends up in the final product, reducing waste disposal costs. The use of a relatively low catalyst loading of 2 mol% further contributes to cost efficiency, making the process economically viable for large-scale commercial production without compromising on yield.
- Enhanced Supply Chain Reliability: The reliance on widely available aromatic aldehydes and 2-aminopyridine ensures that raw material sourcing is not a bottleneck. These commodities are produced by numerous global suppliers, providing procurement teams with multiple sourcing options to mitigate supply risks. The robustness of the reaction conditions, which tolerate a wide range of substituents including halogens and esters, means that the same platform technology can be applied to synthesize a diverse library of analogues without needing to requalify entirely new synthetic routes. This flexibility is invaluable for R&D pipelines where structural modifications are frequent, allowing for rapid scale-up of new candidates with minimal process development time.
- Scalability and Environmental Compliance: Scaling this reaction from gram to kilogram scale is facilitated by the mild thermal requirements and the use of common polar solvents like acetonitrile, which are easy to recover and recycle. The absence of harsh reagents or extreme temperatures reduces the engineering controls required for safe operation, lowering capital expenditure for reactor infrastructure. From an environmental standpoint, the high yield and selectivity minimize the generation of hazardous byproducts, aligning with green chemistry principles and simplifying regulatory compliance for waste management. This sustainable profile is increasingly important for maintaining corporate social responsibility goals and meeting the stringent environmental standards of international pharmaceutical clients.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Rh(III) catalytic technology. These answers are derived directly from the experimental data and technical specifications outlined in the patent documentation, providing a clear understanding of the process capabilities and limitations. Understanding these details is essential for evaluating the feasibility of integrating this method into your existing manufacturing portfolio.
Q: What are the advantages of the in-situ directing group strategy?
A: This strategy eliminates the need for pre-synthesizing complex amide substrates, allowing the use of simple, commercially available aromatic aldehydes and 2-aminopyridine, which significantly simplifies the supply chain and reduces raw material costs.
Q: What is the typical catalyst loading and reaction temperature?
A: The process operates under mild conditions, typically requiring only 2 mol% of [Cp*RhCl2]2 catalyst and a reaction temperature of 80°C, which facilitates easier scale-up and energy efficiency compared to harsher traditional methods.
Q: Is this method suitable for diverse substrate scopes?
A: Yes, the method demonstrates high tolerance for various substituents including alkyl, alkoxy, halogens, and electron-withdrawing groups on both the aromatic aldehyde and the olefin, ensuring broad applicability for generating diverse pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Isoindolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced C-H activation technologies in modern drug discovery and development. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative laboratory methods like the Rh(III) catalyzed synthesis described in CN109232529B can be successfully translated into robust industrial processes. Our state-of-the-art facilities are equipped with rigorous QC labs capable of handling complex nitrogen heterocycles, guaranteeing that every batch meets stringent purity specifications required for clinical and commercial applications. We are committed to delivering high-quality pharmaceutical intermediates that empower our clients to bring life-saving therapies to market faster.
We invite you to explore how this cutting-edge synthesis route can optimize your project economics and accelerate your timeline. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific molecule. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our expertise in Rh-catalyzed transformations can become a cornerstone of your supply chain strategy.