Scaling Multi-Target Kinase Inhibitors: Technical Upgrades and Commercial Mass Production Capabilities
The landscape of oncology drug development is increasingly shifting towards multi-target tyrosine kinase inhibitors that can simultaneously disrupt several signaling pathways involved in tumor proliferation and angiogenesis. A pivotal advancement in this domain is documented in patent CN110028444B, which discloses a novel series of 1-aryl-3-[4-(pyridine-2-yl methoxy) phenyl] urea compounds. These molecules exhibit potent inhibitory activity against critical kinases including BRaf, VEGFR-2, PDGFR-beta, and TOPK, positioning them as high-value candidates for next-generation anti-tumor therapeutics. For pharmaceutical manufacturers and procurement strategists, understanding the synthetic accessibility and structural versatility of these intermediates is crucial for securing a robust supply chain. The patent outlines a chemically elegant route that balances molecular complexity with process efficiency, ensuring that these sophisticated structures can be translated from laboratory discovery to commercial reality without compromising on purity or yield.
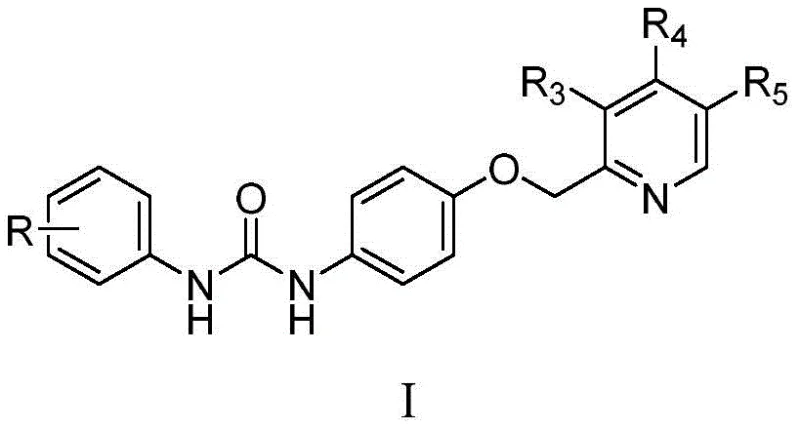
From a strategic sourcing perspective, the ability to access diverse analogs within this chemical class allows R&D teams to optimize pharmacokinetic profiles while maintaining a consistent manufacturing backbone. The general structure permits extensive modification at the aryl and pyridine rings, enabling the fine-tuning of potency against specific cancer cell lines such as A549, HCT-116, and PC3. As a reliable pharmaceutical intermediate supplier, recognizing the nuances of this scaffold is essential for supporting clients who aim to develop broad-spectrum anti-cancer agents. The following analysis dissects the technical merits of this patented technology, highlighting how it addresses common bottlenecks in the synthesis of complex urea derivatives and offering a pathway for cost-effective, scalable production.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the synthesis of diaryl urea derivatives, which serve as the core pharmacophore for many kinase inhibitors, has been plagued by significant operational challenges and safety concerns. Conventional routes often rely on the use of phosgene or its toxic equivalents to generate the urea linkage, necessitating stringent safety protocols and specialized equipment that drive up capital expenditure and operational costs. Furthermore, older methodologies frequently require harsh reaction conditions, such as extreme temperatures or strong acidic environments, which can lead to the degradation of sensitive functional groups present on the pyridine or aryl rings. These aggressive conditions often result in complex impurity profiles that are difficult to purge, requiring extensive and costly downstream purification processes like preparative HPLC. Additionally, the stepwise construction of the ether linkage in prior art often suffers from low conversion rates due to steric hindrance, leading to poor overall yields and increased waste generation. Such inefficiencies create substantial vulnerabilities in the supply chain, causing delays in clinical material production and inflating the cost of goods sold for the final active pharmaceutical ingredient.
The Novel Approach
In contrast, the methodology presented in CN110028444B introduces a streamlined and safer synthetic strategy that circumvents these historical hurdles through intelligent route design. The novel approach utilizes a mild nucleophilic substitution reaction for the etherification step, employing sodium hydroxide in ethanol at room temperature, which eliminates the need for energy-intensive heating or cooling systems. By switching to isocyanates for the final urea coupling step, the process avoids the handling of gaseous phosgene, thereby significantly enhancing workplace safety and reducing regulatory compliance burdens. This route demonstrates exceptional chemoselectivity, preserving the integrity of halogen and alkoxy substituents throughout the synthesis, which is critical for maintaining the biological activity of the final drug substance. The modular nature of this synthesis allows for the rapid generation of a library of analogs by simply varying the starting isocyanate or the chloromethyl pyridine precursor, accelerating the lead optimization phase for drug discovery teams. Ultimately, this technological shift represents a move towards greener chemistry principles, reducing solvent consumption and waste while improving the overall mass balance of the manufacturing process.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The core of this synthetic success lies in the efficient construction of the ether bridge connecting the nitrophenyl and pyridine moieties, a transformation that is mechanistically driven by base-promoted nucleophilic substitution. In this step, the phenolic oxygen of the nitrophenol acts as a nucleophile, attacking the electrophilic chloromethyl carbon of the pyridine derivative. The use of a polar protic solvent like ethanol facilitates the dissolution of the inorganic base and stabilizes the transition state, ensuring high conversion rates even at ambient temperatures. This is followed by a catalytic hydrogenation step using palladium on carbon, which selectively reduces the nitro group to an aniline without affecting the newly formed ether bond or other sensitive substituents like halogens. The final urea formation proceeds via a nucleophilic attack of the aniline nitrogen on the electrophilic carbon of the isocyanate group, a reaction that is thermodynamically favorable and proceeds rapidly in dichloromethane. This sequence ensures that the most reactive functionalities are introduced at the optimal stage of the synthesis, minimizing side reactions and maximizing the yield of the desired regioisomer.
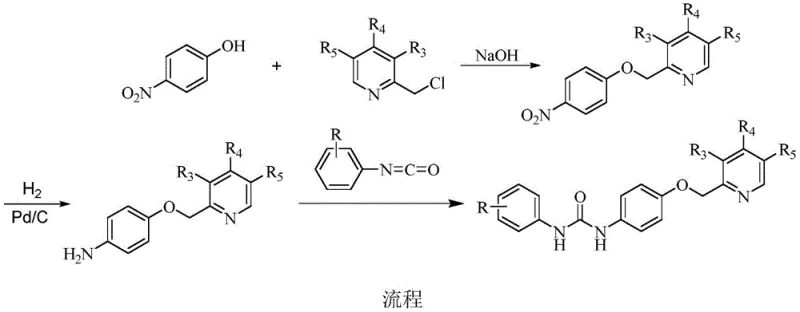
Impurity control is rigorously managed through the selection of high-purity starting materials and the implementation of straightforward workup procedures such as filtration and crystallization. For instance, the reduction step yields a pale yellow oily substance that can be used directly in the next step without further purification, demonstrating the robustness of the reaction against minor byproducts. The final urea coupling is monitored to ensure complete consumption of the isocyanate, preventing the presence of toxic residual reagents in the final product. By understanding these mechanistic details, process chemists can better anticipate potential failure modes and implement proactive quality control measures. This depth of technical understanding is vital for ensuring batch-to-batch consistency, which is a non-negotiable requirement for regulatory filings and commercial supply agreements in the pharmaceutical industry.
How to Synthesize 1-Aryl-3-Urea Compounds Efficiently
To achieve optimal results in the production of these kinase inhibitor intermediates, operators must adhere to precise stoichiometric ratios and maintain strict control over reaction parameters such as temperature and addition rates. The patented procedure outlines a clear sequence beginning with the etherification of nitrophenol, followed by catalytic reduction, and concluding with the urea coupling reaction. Each step has been optimized to balance reaction speed with product quality, ensuring that the process is both time-efficient and capable of delivering high-purity materials suitable for biological testing. Detailed standard operating procedures regarding solvent drying, catalyst loading, and quenching protocols are essential for replicating the high yields reported in the patent examples.
- Perform nucleophilic substitution between nitrophenol and chloromethyl pyridine derivatives using sodium hydroxide in ethanol.
- Execute catalytic hydrogenation using Pd/C to reduce the nitro group to an amine under ambient pressure.
- React the resulting aniline derivative with substituted phenyl isocyanates in dichloromethane to form the final urea linkage.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers tangible benefits that extend beyond mere technical feasibility, directly impacting the bottom line and operational resilience. The elimination of hazardous reagents like phosgene reduces the need for specialized containment infrastructure, allowing production to be conducted in standard multipurpose reactors found in most fine chemical facilities. This flexibility significantly lowers the barrier to entry for manufacturing partners and increases the number of qualified suppliers available in the market, thereby mitigating single-source risks. Furthermore, the use of commodity chemicals such as ethanol, methanol, and sodium hydroxide ensures that raw material costs remain stable and predictable, shielding the project from volatile pricing associated with exotic reagents. The high yields observed in key steps, particularly the reduction phase, mean that less starting material is required to produce a given amount of final product, directly translating to substantial cost savings in raw material procurement.
- Cost Reduction in Manufacturing: The process achieves significant economic efficiency by utilizing inexpensive, readily available solvents and reagents that do not require complex storage or handling protocols. By avoiding cryogenic conditions and high-pressure reactors, the energy consumption per kilogram of product is drastically reduced, leading to lower utility costs. The ability to telescope certain steps or use crude intermediates without purification further minimizes processing time and labor expenses. Additionally, the high selectivity of the reactions reduces the burden on waste treatment facilities, lowering environmental compliance costs associated with hazardous waste disposal. These cumulative factors contribute to a leaner cost structure that enhances the competitiveness of the final drug product in price-sensitive markets.
- Enhanced Supply Chain Reliability: The reliance on bulk commodity chemicals ensures that the supply chain is robust against disruptions that might affect niche or specialty reagents. Since the starting materials like nitrophenols and chloromethyl pyridines are produced by multiple global vendors, procurement teams can easily qualify alternative sources to prevent bottlenecks. The simplicity of the synthesis also means that technology transfer to contract manufacturing organizations is faster and less prone to errors, ensuring consistent supply continuity during scale-up. This reliability is critical for maintaining clinical trial timelines and ensuring that commercial launch dates are met without delay due to material shortages. Consequently, partners can plan their inventory levels with greater confidence, reducing the need for excessive safety stock.
- Scalability and Environmental Compliance: The reaction conditions are inherently scalable, having been demonstrated to work effectively without the need for specialized equipment that is difficult to replicate at large volumes. The use of aqueous workups and simple filtration steps aligns well with green chemistry initiatives, reducing the volume of organic solvent waste generated per unit of product. This environmental friendliness simplifies the permitting process for new manufacturing sites and aligns with the sustainability goals of major pharmaceutical companies. The process generates minimal hazardous byproducts, making waste treatment more straightforward and less costly. Overall, the route supports a sustainable manufacturing model that can grow from pilot plant quantities to multi-ton commercial production without fundamental changes to the chemistry.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these 1-aryl-3-urea compounds. These answers are derived directly from the experimental data and specifications outlined in the patent documentation, providing clarity on performance metrics and process capabilities. Understanding these details helps stakeholders make informed decisions regarding the integration of this technology into their development pipelines. The responses cover aspects ranging from biological activity to manufacturing feasibility, ensuring a comprehensive overview of the value proposition.
Q: What are the primary biological targets of these 1-aryl-3-urea compounds?
A: These compounds function as multi-target inhibitors specifically designed to block BRaf kinase, VEGFR-2, PDGFR-beta, and TOPK, offering a comprehensive approach to inhibiting tumor signal transduction and angiogenesis.
Q: How does this synthesis route improve upon conventional urea formation methods?
A: The patented method avoids the use of hazardous phosgene equivalents by utilizing stable isocyanates for coupling, and employs mild room-temperature conditions for etherification, significantly enhancing operational safety and scalability.
Q: Is the process suitable for large-scale commercial production?
A: Yes, the synthesis utilizes common solvents like ethanol and methanol, avoids cryogenic conditions, and features high-yield steps such as the 94.1% yield in the reduction phase, making it highly adaptable for industrial tonnage production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 1-Aryl-3-Urea Compounds Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of having a manufacturing partner who can navigate the complexities of synthesizing multi-target kinase inhibitors with precision and reliability. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can seamlessly transition from preclinical research to full-scale market supply. We are committed to meeting stringent purity specifications through our rigorous QC labs, which employ advanced analytical techniques to verify the identity and quality of every batch. Our facility is equipped to handle the specific requirements of urea chemistry, including the safe management of isocyanates and the efficient execution of catalytic hydrogenations. By leveraging our technical expertise, you can accelerate your development timeline and reduce the risks associated with process scale-up.
We invite you to engage with our technical procurement team to discuss how we can support your specific needs for high-purity pharmaceutical intermediates. Whether you require a Customized Cost-Saving Analysis for your current supply chain or need to evaluate the feasibility of a new analog, we are ready to provide detailed assessments. Please contact us to request specific COA data and route feasibility assessments tailored to your project milestones. Our goal is to become an extension of your R&D and supply chain operations, delivering value through technical excellence and unwavering commitment to quality.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →