Advanced Synthesis of Fluoroquinolone Dimer Derivatives for Oncology Applications
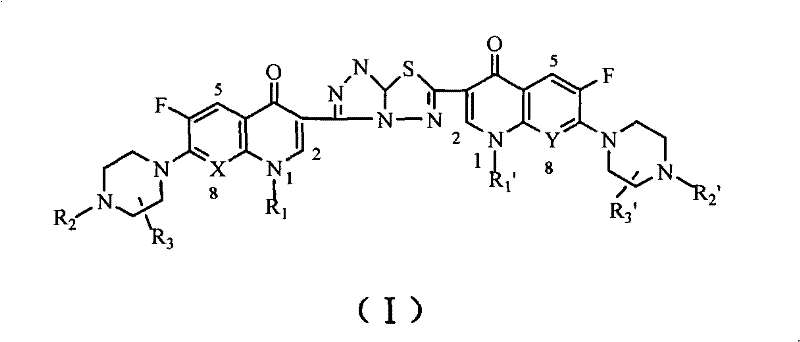
The pharmaceutical landscape is continuously evolving towards multi-target therapies, and patent CN101648962B introduces a significant breakthrough in this domain with the disclosure of C3/C3 fluoroquinolone dimer derivatives linked by an s-triazolo[3,4-b][1,3,4]thiadiazole chain. This innovation represents a strategic shift from traditional antibacterial applications of fluoroquinolones to potent antitumor agents, leveraging the structural homology between bacterial topoisomerase and mammalian topoisomerase II. The core invention focuses on modifying the C-3 carboxyl group, traditionally considered essential for antibacterial activity but dispensable for antitumor effects, to create a dimeric architecture that exhibits enhanced cytotoxicity against leukemia cell lines such as HL60 and L1210. This structural modification not only preserves the inherent antibacterial backbone but also introduces a new mechanism of action that addresses the common limitations of low bioavailability and rapid metabolic deactivation found in earlier generations of quinolone-based anticancer candidates. For R&D directors seeking novel scaffolds, this technology offers a robust platform for developing high-purity pharmaceutical intermediates with dual therapeutic potential.
The limitations of conventional methods for developing antitumor fluoroquinolones often stem from the direct modification of the pharmacophore, which can lead to a loss of potency or increased toxicity profiles that parallel the desired activity. Traditional approaches involving bicyclic or tricyclic quinolone modifications have struggled with poor aqueous solubility and instability in physiological conditions, resulting in suboptimal pharmacokinetic profiles that hinder clinical progression. Furthermore, monomeric fluoroquinolone derivatives frequently suffer from rapid clearance and metabolic inactivation in vivo, necessitating high dosing regimens that increase the risk of adverse effects. The reliance on single-molecule architectures also limits the ability to engage multiple biological targets simultaneously, which is increasingly recognized as a critical factor in overcoming drug resistance in oncology. These challenges underscore the need for a more sophisticated molecular design that can enhance stability and potency without compromising the safety profile established by decades of fluoroquinolone clinical use.
The novel approach detailed in the patent overcomes these hurdles by employing a dimerization strategy that links two fluoroquinolone units through a stable s-triazolo[3,4-b][1,3,4]thiadiazole bridge at the C3 position. This specific linker is not merely a spacer but an active pharmacophore contributor that enhances the overall electron distribution and steric fit within the topoisomerase binding pocket. By utilizing readily available fluoroquinolones such as norfloxacin, ciprofloxacin, and levofloxacin as starting materials, the synthesis ensures that the core quinolone integrity is maintained while the dimeric structure introduces synergistic effects. The method avoids the use of rare or prohibitively expensive catalysts, relying instead on standard industrial reagents like hydrazine hydrate and carbon disulfide, which facilitates a smoother transition from laboratory scale to commercial production. This design effectively transforms a well-understood class of antibiotics into a new class of antitumor agents with improved therapeutic indices and reduced likelihood of resistance development.
Mechanistic Insights into s-triazolo[3,4-b][1,3,4]thiadiazole Linker Formation
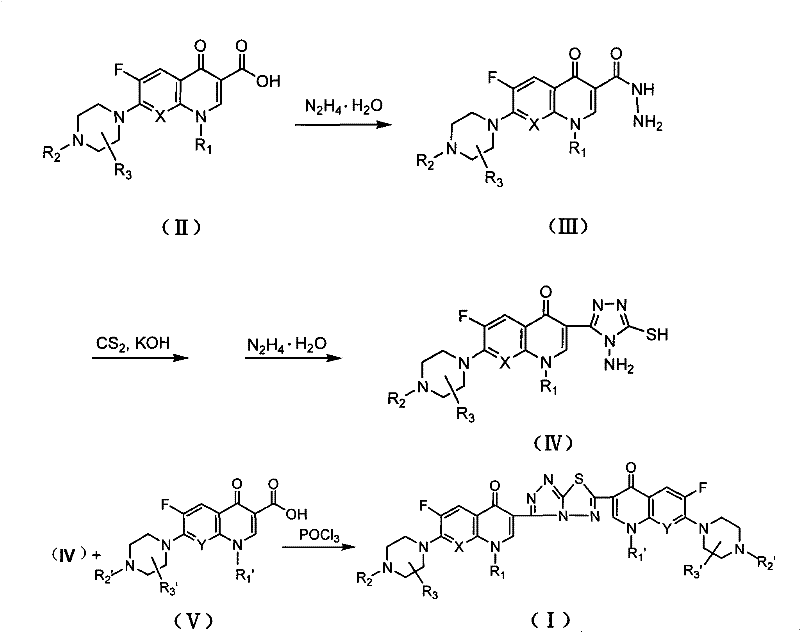
The mechanistic pathway for constructing the s-triazolo[3,4-b][1,3,4]thiadiazole linker involves a precise sequence of heterocyclic cyclization reactions that ensure high regioselectivity and purity. The process begins with the conversion of the fluoroquinolone-3-carboxylic acid into an acyl hydrazide through nucleophilic attack by hydrazine hydrate under reflux conditions, a transformation that activates the C3 position for subsequent ring closure. This intermediate is then subjected to cyclization with carbon disulfide in the presence of potassium hydroxide, forming a potassium dithiocarbazinate salt which subsequently undergoes intramolecular condensation to yield the 1-amino-5-mercapto-1,2,4-triazole core. The final step involves the condensation of this triazole intermediate with a second molecule of fluoroquinolone-3-carboxylic acid in phosphorus oxychloride, which acts as both a solvent and a dehydrating agent to drive the formation of the fused thiadiazole ring system. This cascade of reactions is highly efficient and minimizes the formation of side products, ensuring that the final dimeric structure possesses the precise stereochemistry required for optimal biological activity.
Impurity control in this synthesis is critical due to the complexity of the dimeric structure and the potential for incomplete cyclization or hydrolysis of the sensitive heterocyclic linker. The protocol specifies rigorous purification steps, including recrystallization from ethanol and pH-adjusted precipitation, to remove unreacted starting materials and intermediate byproducts such as uncondensed hydrazides or open-chain triazoles. The use of phosphorus oxychloride in the final step requires careful quenching and neutralization to prevent the formation of chlorinated impurities that could compromise the safety profile of the final pharmaceutical intermediate. Analytical data from the patent indicates that the resulting compounds exhibit sharp melting points and consistent mass spectrometry profiles, confirming the high degree of chemical homogeneity achieved through this method. For quality assurance teams, this robust impurity profile simplifies the regulatory filing process and ensures that the material meets the stringent specifications required for clinical-grade active pharmaceutical ingredients.
How to Synthesize Fluoroquinolone Dimer Derivatives Efficiently
The synthesis of these high-value dimeric compounds follows a standardized three-step protocol that is amenable to scale-up in a GMP-compliant manufacturing environment. The process begins with the activation of the fluoroquinolone acid, followed by the construction of the heterocyclic bridge, and concludes with the coupling of the second quinolone unit to form the final dimer. Detailed standardized synthesis steps are provided in the guide below to ensure reproducibility and yield optimization across different production batches. This streamlined approach minimizes unit operations and reduces the overall processing time, making it an attractive candidate for contract development and manufacturing organizations looking to expand their oncology pipeline.
- React fluoroquinolone carboxylic acid with excess hydrazine hydrate under reflux to form the acyl hydrazide intermediate.
- Cyclize the hydrazide with carbon disulfide and potassium hydroxide, followed by hydrazine treatment to generate the amino-mercapto-triazole.
- Condense the triazole intermediate with a second fluoroquinolone molecule in phosphorus oxychloride to yield the final dimer.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, this synthesis route offers substantial cost reduction in pharmaceutical intermediates manufacturing by utilizing bulk commodity chemicals as the primary starting materials. The reliance on established fluoroquinolones like ciprofloxacin and norfloxacin means that the raw material supply chain is mature, stable, and less susceptible to the volatility often associated with exotic or custom-synthesized building blocks. The elimination of precious metal catalysts or complex chiral auxiliaries further drives down the bill of materials, allowing for a more competitive pricing structure without sacrificing quality. Additionally, the high yields reported in the patent examples suggest that material throughput is efficient, reducing the waste disposal costs and environmental footprint associated with low-yielding synthetic pathways. This economic efficiency translates directly into better margin protection for downstream drug developers and ensures a reliable supply of critical intermediates for long-term clinical programs.
- Cost Reduction in Manufacturing: The synthesis strategy significantly lowers production costs by avoiding the use of expensive transition metal catalysts and specialized reagents that typically drive up the price of complex heterocyclic compounds. By leveraging common industrial solvents and reagents such as hydrazine hydrate and carbon disulfide, the process minimizes the need for costly purification steps like chromatography, relying instead on crystallization and precipitation which are far more scalable and economical. The atom economy of the dimerization step is also favorable, as the phosphorus oxychloride mediated condensation efficiently joins two large molecular fragments with minimal loss of mass. These factors combine to create a manufacturing process that is inherently lean, reducing the overall cost of goods sold and enabling more aggressive pricing strategies in the competitive oncology intermediate market.
- Enhanced Supply Chain Reliability: Supply chain continuity is bolstered by the use of widely available starting materials that are produced by multiple global suppliers, mitigating the risk of single-source bottlenecks. The synthetic route does not depend on temperature-sensitive biologics or unstable reagents that require cold-chain logistics, simplifying storage and transportation requirements for raw materials. Furthermore, the robustness of the reaction conditions, which tolerate standard industrial equipment and ambient pressure, ensures that production can be maintained even during periods of supply chain disruption or equipment maintenance. This resilience is crucial for maintaining consistent delivery schedules to pharmaceutical clients who rely on just-in-time inventory models for their clinical trial materials and commercial launches.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing reaction conditions that can be safely transferred from laboratory glassware to large-scale stainless steel reactors without significant re-optimization. The waste streams generated are primarily aqueous and organic solvents that can be treated using standard effluent treatment protocols, ensuring compliance with increasingly stringent environmental regulations. The absence of heavy metal residues in the final product eliminates the need for expensive metal scavenging steps, further simplifying the downstream processing and reducing the environmental impact of the manufacturing operation. This alignment with green chemistry principles not only reduces regulatory risk but also enhances the corporate social responsibility profile of the supply chain, appealing to environmentally conscious stakeholders and investors.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these fluoroquinolone dimer derivatives. The answers are derived directly from the patent specifications and experimental data to provide accurate guidance for potential partners and licensees. Understanding these details is essential for evaluating the feasibility of integrating this technology into existing drug development pipelines.
Q: What is the primary advantage of the s-triazolo linker in fluoroquinolone dimers?
A: The s-triazolo[3,4-b][1,3,4]thiadiazole linker connects two fluoroquinolone units at the C3 position, enhancing cytotoxic activity against leukemia cell lines compared to monomeric forms while maintaining antibacterial properties.
Q: Are the starting materials for this synthesis commercially available?
A: Yes, the synthesis utilizes established fluoroquinolones such as norfloxacin, ciprofloxacin, and levofloxacin as starting materials, which are widely available and cost-effective bulk chemicals.
Q: How does this method address solubility issues common in quinolone drugs?
A: By modifying the C3 carboxyl group into a dimeric structure via a heterocyclic linker, the derivative alters the physicochemical properties, potentially improving bioavailability and overcoming metabolic deactivation seen in traditional monomers.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Fluoroquinolone Dimer Derivatives Supplier
NINGBO INNO PHARMCHEM stands at the forefront of custom synthesis for complex pharmaceutical intermediates, possessing the technical expertise to scale diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facility is equipped with rigorous QC labs and stringent purity specifications to ensure that every batch of fluoroquinolone dimer derivatives meets the highest international standards for oncology research. We understand the critical nature of supply chain stability in drug development and are committed to providing a seamless partnership that supports your timeline from preclinical studies through to commercial launch. Our team of chemists is ready to optimize this specific dimerization route to maximize yield and minimize impurities tailored to your specific process requirements.
We invite you to contact our technical procurement team to request specific COA data and route feasibility assessments for your project. By collaborating with us, you gain access to a Customized Cost-Saving Analysis that identifies opportunities to further optimize the manufacturing economics of your target molecule. Let us help you accelerate your development timeline with a reliable supply of high-quality intermediates that empower your research and ensure the success of your therapeutic candidates.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →