Advanced Synthesis and Commercial Scale-Up of Next-Generation Pirfenidone Derivatives
The global demand for effective treatments against Idiopathic Pulmonary Fibrosis (IPF) continues to drive intense research into next-generation therapeutics that overcome the limitations of current clinical standards. Patent CN114057630B discloses a groundbreaking class of pirfenidone derivatives designed to address the critical issue of low bioavailability and metabolic instability associated with the parent drug. By strategically replacing the metabolically labile 5-methyl group with a stable amide linkage, this technology offers a robust platform for developing superior anti-fibrotic agents. As a leading manufacturer in the fine chemical sector, we recognize the immense potential of this synthetic pathway to deliver high-value pharmaceutical intermediates to the global market. The following technical analysis details the mechanistic elegance and commercial viability of this novel approach.
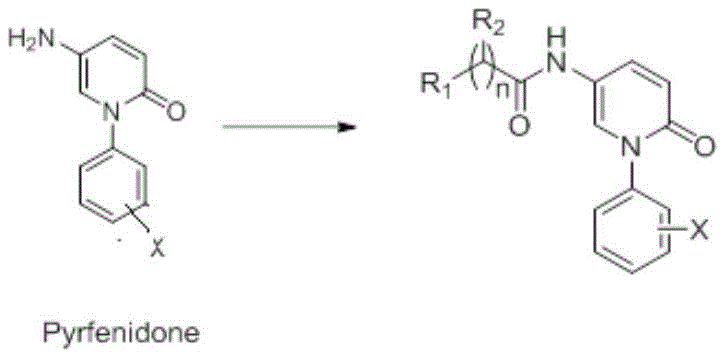
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional pirfenidone synthesis and its subsequent modification often struggle with metabolic liabilities that hinder therapeutic efficacy. The native 5-methyl group on the pyridinone ring is prone to rapid oxidation by hepatic enzymes into hydroxymethyl or carboxyl metabolites, which are pharmacologically inactive. This metabolic clearance necessitates high daily dosages in clinical settings, directly correlating with increased incidence of adverse effects such as gastrointestinal distress, photosensitivity, and nausea. Furthermore, conventional functionalization strategies at the 5-position frequently rely on harsh conditions or multi-step sequences that suffer from poor atom economy and difficult purification profiles. These factors collectively create a bottleneck in the cost reduction in anti-fibrosis drug manufacturing, as the yield losses and extensive downstream processing inflate the cost of goods significantly.
The Novel Approach
The methodology outlined in the patent introduces a paradigm shift by utilizing a hypervalent iodine-mediated C-N coupling strategy to construct the core scaffold efficiently. Instead of struggling with direct alkylation, the process builds the molecule through a convergent route starting from 2-hydroxy-5-nitropyridine. This approach allows for the precise installation of diverse amide side chains, enabling the hybridization of pirfenidone with known anti-inflammatory pharmacophores. The result is a library of compounds where the amide bond provides metabolic stability while the variable R-groups tune the biological activity. This modularity is crucial for a reliable pharmaceutical intermediate supplier aiming to support diverse drug discovery programs. The visual representation of this streamlined synthesis highlights the logical progression from simple precursors to complex, bioactive targets.
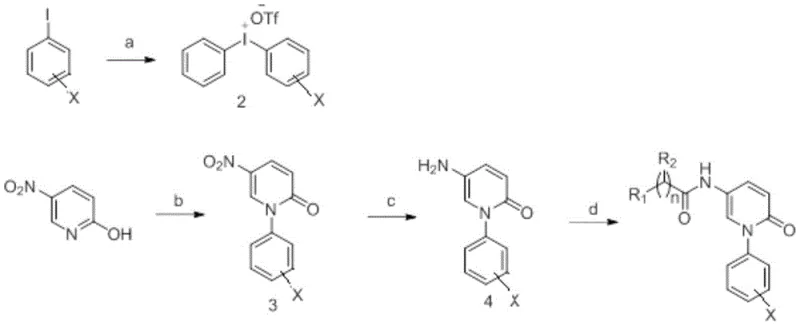
Mechanistic Insights into Hypervalent Iodine-Mediated C-N Coupling
The cornerstone of this synthetic innovation is the generation and utilization of a diaryliodonium salt intermediate. The process begins with the oxidation of iodobenzene using meta-chloroperbenzoic acid (m-CPBA) in the presence of trifluoromethanesulfonic acid (TfOH) to generate a highly reactive hypervalent iodine species. This electrophilic iodine center then facilitates a copper-catalyzed C-N coupling with 2-hydroxy-5-nitropyridine. Unlike traditional palladium-catalyzed cross-couplings which can be sensitive to moisture and require expensive ligands, this copper-mediated protocol operates under relatively mild conditions, typically in dichloromethane with triethylamine as a base. The mechanism likely involves the coordination of the pyridone nitrogen to the copper center, followed by oxidative addition or ligand exchange with the iodonium salt, ultimately forging the N-aryl bond with high regioselectivity. This step is critical for establishing the high-purity pirfenidone derivatives required for biological testing, as it minimizes the formation of homocoupling byproducts often seen in radical-based arylation methods.
Following the coupling, the nitro group serves as a versatile handle for further functionalization. The reduction step employs iron powder and ammonium chloride in an ethanol-water mixture, a classic yet highly effective method for converting nitroarenes to anilines. This choice of reductant is commercially advantageous as it avoids the use of hydrogen gas and high-pressure equipment, simplifying the safety profile for commercial scale-up of complex heterocyclic intermediates. The resulting amine intermediate is then subjected to amide bond formation using standard carbodiimide chemistry, specifically EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide) and HOBt (1-hydroxybenzotriazole). This activation strategy ensures efficient coupling with a wide range of carboxylic acids, from simple benzoic acids to complex bicyclic systems, without racemization of chiral centers if present. The robustness of this final amidation step allows for the rapid generation of large compound libraries, accelerating the structure-activity relationship (SAR) studies essential for modern drug development.
How to Synthesize Pirfenidone Derivatives Efficiently
The synthesis of these targeted anti-fibrotic agents follows a logical four-step sequence that balances chemical efficiency with operational simplicity. Beginning with the preparation of the hypervalent iodine coupling partner, the route proceeds through C-N bond formation, nitro reduction, and final amide condensation. Each step has been optimized to maximize yield and minimize impurity carryover, ensuring that the final active pharmaceutical ingredient (API) precursors meet stringent quality standards. For process chemists looking to implement this workflow, the detailed standardized synthesis steps are provided in the guide below to ensure reproducibility and safety.
- Preparation of hypervalent iodine salt from iodobenzene and benzene using m-CPBA and triflic acid.
- C-N coupling of 2-hydroxy-5-nitropyridine with the iodine salt using CuI catalysis to form the nitro-intermediate.
- Reduction of the nitro group to an amine using iron powder and ammonium chloride, followed by amide formation with carboxylic acids.
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain perspective, the adoption of this synthetic route offers substantial strategic benefits over legacy methods. The reliance on commodity chemicals such as iodobenzene, benzene, and iron powder ensures a stable and resilient raw material supply base, mitigating the risks associated with sourcing exotic or single-source reagents. This stability is paramount for maintaining continuous production schedules and reducing lead time for high-purity API precursors. Furthermore, the avoidance of precious metal catalysts like palladium in the key coupling step eliminates the need for costly metal scavenging processes and rigorous residual metal testing, which are significant cost drivers in GMP manufacturing. The purification methods described, primarily involving recrystallization and standard column chromatography, are easily transferable from laboratory to pilot plant scales, facilitating a smoother technology transfer process.
- Cost Reduction in Manufacturing: The synthetic pathway is designed to maximize atom economy and minimize waste generation. By achieving a total yield above 60% across four steps, the process significantly reduces the consumption of starting materials per kilogram of final product. The use of inexpensive reagents like iron powder for reduction and EDC/HOBt for amidation, rather than catalytic hydrogenation or specialized coupling reagents, drives down the direct material costs. Additionally, the simplified workup procedures reduce solvent usage and energy consumption, contributing to a lower overall carbon footprint and operational expenditure.
- Enhanced Supply Chain Reliability: The modular nature of the synthesis allows for flexible manufacturing strategies. The key nitro-intermediate can be produced in bulk and stored, serving as a common precursor for multiple final derivatives. This decoupling of the upstream and downstream processes enhances supply chain agility, allowing manufacturers to respond quickly to fluctuating demand for specific analogs. The robustness of the reaction conditions also means that production is less susceptible to minor variations in raw material quality or environmental conditions, ensuring consistent delivery performance to downstream partners.
- Scalability and Environmental Compliance: The process avoids the use of highly toxic reagents or extreme reaction conditions that would complicate regulatory approval and environmental permitting. The aqueous workup in the reduction step and the use of common organic solvents like dichloromethane and ethyl acetate align with standard industrial waste treatment protocols. This compliance readiness accelerates the timeline for regulatory filings and commercial launch. Moreover, the high purity of the crude products obtained through recrystallization minimizes the need for extensive chromatographic purification at scale, further enhancing the environmental sustainability and economic feasibility of the manufacturing process.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these pirfenidone derivatives. These answers are derived directly from the experimental data and technical specifications provided in the patent literature, ensuring accuracy and relevance for industry stakeholders. Understanding these details is crucial for evaluating the feasibility of integrating these intermediates into your drug development pipeline.
Q: What is the key structural modification in these new pirfenidone derivatives?
A: The 5-methyl group of traditional pirfenidone is replaced with a metabolically stable amide group, often hybridized with anti-inflammatory moieties like aspirin or ursolic acid fragments, to improve bioavailability and reduce side effects.
Q: How does the synthesis route ensure scalability for industrial production?
A: The route utilizes readily available starting materials like iodobenzene and 2-hydroxy-5-nitropyridine, avoids expensive precious metal catalysts in the final steps, and employs standard purification techniques like recrystallization, achieving a total yield above 60%.
Q: What is the biological activity profile of these compounds compared to clinical pirfenidone?
A: In vitro screening on NIH3T3 fibroblast cells demonstrates that most derivatives exhibit superior anti-proliferative activity, with lead compounds showing significantly enhanced potency, making them promising candidates for idiopathic pulmonary fibrosis treatment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Pirfenidone Derivatives Supplier
The technological advancements presented in patent CN114057630B represent a significant leap forward in the fight against pulmonary fibrosis, offering a clear path to more effective and safer medicines. At NINGBO INNO PHARMCHEM, we possess the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring these complex molecules from the bench to the clinic. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, ensuring that every batch of intermediate we deliver supports your critical research and development goals with unwavering reliability.
We invite you to collaborate with us to optimize your supply chain for these high-value anti-fibrosis agents. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality needs. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how our manufacturing expertise can accelerate your project timelines and enhance your competitive edge in the pharmaceutical market.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →