Advanced Manufacturing of Olmesartan Intermediates: Technical Breakthroughs and Commercial Scalability
Advanced Manufacturing of Olmesartan Intermediates: Technical Breakthroughs and Commercial Scalability
The pharmaceutical landscape for Angiotensin II Receptor Blockers (ARBs) continues to evolve, driven by the relentless demand for higher purity active pharmaceutical ingredients (APIs) and more efficient manufacturing pathways. Patent CN1532195A introduces a transformative methodology for the preparation of Olmesartan, a potent antihypertensive agent, addressing long-standing challenges in synthetic efficiency and impurity control. This technical disclosure outlines a robust sequence involving the strategic ring-opening of a furo-imidazolone precursor, followed by selective esterification and final deprotection. For R&D directors and process chemists, this route represents a significant departure from traditional etherification-heavy protocols, offering a cleaner reaction profile that minimizes the formation of difficult-to-remove by-products. By shifting the synthetic logic towards a hydrolysis-first approach, the process achieves superior selectivity, ensuring that the critical tetrazole and biphenyl motifs remain intact while the carboxylic acid functionality is precisely manipulated for final assembly.
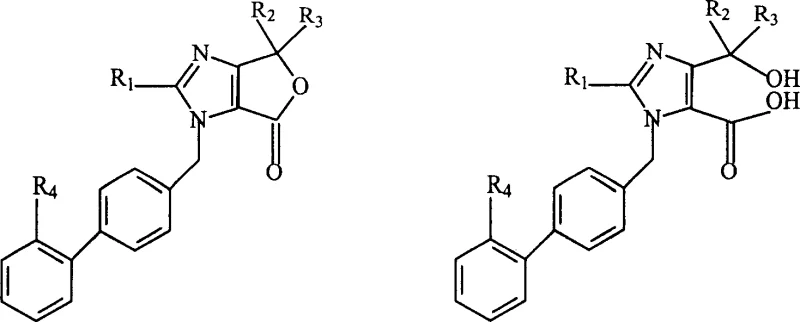
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Olmesartan and its precursors has been plagued by inherent chemoselectivity issues, particularly during the coupling of the imidazole core with the biphenyl-tetrazole side chain. Traditional routes often rely on direct etherification strategies where the 4-hydroxyl group of the imidazole intermediate is susceptible to competing reactions with the electrophilic sites on the biphenyl portion. This lack of orthogonality leads to the generation of structurally similar impurities that are notoriously difficult to separate via standard crystallization or chromatography techniques. Furthermore, these conventional pathways frequently necessitate harsh reaction conditions or expensive coupling agents to drive the equilibrium forward, resulting in suboptimal yields and increased waste generation. For procurement managers, these inefficiencies translate directly into higher raw material costs and extended production cycles, as multiple purification steps are required to meet the stringent purity specifications demanded by regulatory bodies for cardiovascular medications.
The Novel Approach
In stark contrast, the methodology disclosed in CN1532195A employs a clever retrosynthetic disconnection that prioritizes the stability of the core scaffold before final functionalization. The process initiates with the alkaline or acidic hydrolysis of a cyclic furo-imidazolone derivative, effectively unmasking the carboxylic acid handle without disturbing the sensitive tetrazole protection. This open-chain intermediate is then subjected to a mild esterification with a specific dioxolone derivative, a step that proceeds with high fidelity under basic catalysis. By decoupling the ring-opening from the final coupling event, the inventors have successfully eliminated the primary source of etherification by-products. This strategic reordering of synthetic steps not only streamlines the workflow but also significantly enhances the overall mass balance of the process, making it an attractive candidate for cost reduction in pharmaceutical intermediate manufacturing where margin compression is a constant pressure.
Mechanistic Insights into Base-Catalyzed Esterification and Deprotection
The core of this synthetic innovation lies in the precise control of nucleophilicity during the esterification phase. When the carboxylic acid intermediate, generated from the ring-opening of the furo-imidazolone, is treated with a base such as potassium carbonate in a polar aprotic solvent like DMF, it forms a reactive carboxylate anion. This anion acts as a potent nucleophile, attacking the electrophilic methylene carbon of the 4-bromomethyl-5-methyl-2-oxo-1,3-dioxole. The mechanistic advantage here is twofold: first, the steric environment of the imidazole carboxylate favors attack at the primary halide over other potential sites; second, the mild basic conditions prevent the degradation of the acid-labile trityl protecting group on the tetrazole ring, which is crucial for maintaining the integrity of the molecule until the final step. This level of chemoselectivity is paramount for ensuring a clean impurity profile, as it avoids the scrambling of protecting groups that often complicates ARB synthesis.
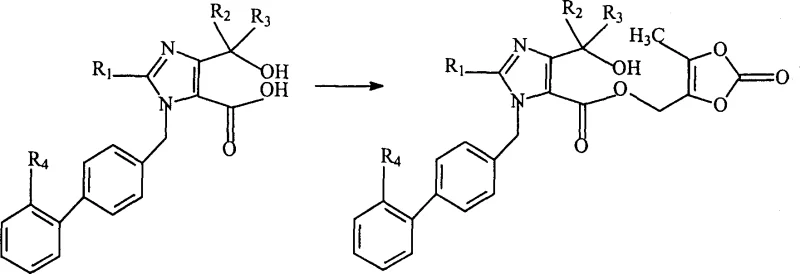
Following the successful formation of the ester linkage, the final transformation involves the removal of the trityl group to reveal the active tetrazole moiety. This deprotection is elegantly achieved using common mineral or organic acids, such as acetic acid or hydrochloric acid, in aqueous or mixed solvent systems. The mechanism proceeds via protonation of the trityl group, facilitating the formation of a stable trityl cation and releasing the free tetrazole nitrogen. The choice of acid and solvent is critical here; the patent suggests that acetic acid provides an optimal balance of solubility and reactivity, allowing the reaction to proceed at moderate temperatures (around 45°C) without inducing hydrolysis of the newly formed ester bond. This orthogonal deprotection strategy ensures that the final Olmesartan molecule is obtained in high purity, with the removal of the trityl by-product being straightforward through filtration or extraction, thereby simplifying the isolation protocol for supply chain teams.
How to Synthesize Olmesartan Efficiently
Implementing this synthesis route requires careful attention to solvent selection and stoichiometry to maximize yield and minimize waste. The process begins with the hydrolysis of the cyclic precursor, where controlling the pH and temperature is essential to ensure complete ring opening without degrading the biphenyl scaffold. Once the carboxylic acid intermediate is isolated, the subsequent esterification must be performed under anhydrous conditions to prevent hydrolysis of the alkylating agent. Finally, the deprotection step demands precise monitoring to ensure complete removal of the trityl group while preserving the ester linkage. For detailed operational parameters, including specific molar ratios, solvent volumes, and work-up procedures, please refer to the standardized synthesis guide below which encapsulates the critical process parameters derived from the patent examples.
- Hydrolyze the furo-imidazolone precursor (Compound A) using alkali or acid in a solvent system like acetone/water to obtain the open-chain carboxylic acid intermediate.
- React the resulting carboxylic acid with 4-bromomethyl-5-methyl-2-oxo-1,3-dioxole under basic catalysis in an inert solvent such as DMF to form the ester linkage.
- Remove the trityl protecting group from the tetrazole ring using an organic or mineral acid, such as acetic acid or hydrochloric acid, to yield the final Olmesartan product.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthetic route offers compelling advantages that resonate deeply with procurement strategies focused on cost efficiency and supply continuity. The primary value driver is the substantial reduction in processing complexity; by eliminating the need for extensive chromatographic purification to remove etherification by-products, manufacturers can significantly lower their operational expenditures. This simplification of the downstream processing train means that existing equipment can be utilized more effectively, increasing batch turnover rates and reducing the overall manufacturing lead time. For supply chain heads, this translates into a more responsive production capability, allowing for quicker adjustments to market demand fluctuations without the bottleneck of lengthy purification cycles that typically characterize complex ARB synthesis.
- Cost Reduction in Manufacturing: The economic benefits of this process are derived principally from the improved atom economy and the elimination of expensive purification media. By avoiding the formation of intractable by-products, the yield of the desired intermediate is inherently higher, meaning less raw material is wasted per kilogram of final product. Furthermore, the reagents employed—such as sodium hydroxide, potassium carbonate, and acetic acid—are commodity chemicals with stable pricing and widespread availability, shielding the supply chain from the volatility associated with specialized catalysts or exotic reagents. This reliance on bulk chemicals ensures that the cost of goods sold (COGS) remains competitive, providing a buffer against margin erosion in the generic pharmaceutical market.
- Enhanced Supply Chain Reliability: Supply security is bolstered by the robustness of the reaction conditions, which tolerate a wider range of operating parameters compared to sensitive transition-metal catalyzed alternatives. The use of common solvents like acetone, ethanol, and DMF ensures that solvent supply disruptions are unlikely to halt production. Additionally, the high selectivity of the reaction reduces the risk of batch failures due to impurity spikes, a common cause of supply interruptions in fine chemical manufacturing. This reliability allows for more accurate forecasting and inventory planning, ensuring that critical API intermediates are available when needed to support downstream drug formulation schedules.
- Scalability and Environmental Compliance: The pathway is designed with industrial scale-up in mind, utilizing reaction conditions that are easily transferable from laboratory to pilot and commercial scales. The absence of heavy metal catalysts simplifies the environmental compliance landscape, reducing the burden of wastewater treatment and hazardous waste disposal. This "green chemistry" aspect not only lowers disposal costs but also aligns with the increasingly stringent environmental regulations faced by chemical manufacturers globally. The ability to scale this process to multi-ton quantities without compromising quality makes it an ideal solution for meeting the growing global demand for antihypertensive therapies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Olmesartan synthesis route. These insights are derived directly from the experimental data and claims presented in the patent literature, providing a factual basis for evaluating the technology's fit within your existing manufacturing portfolio. Understanding these nuances is essential for making informed decisions about process adoption and supplier qualification.
Q: How does this new process improve upon conventional Olmesartan synthesis methods?
A: Conventional methods often suffer from poor selectivity during the etherification step, leading to significant by-product formation where the 4-hydroxyl group reacts with the biphenyl moiety. This novel approach circumvents those issues by utilizing a ring-opening strategy followed by controlled esterification, drastically reducing side reactions and simplifying downstream purification.
Q: What are the critical reaction conditions for the esterification step?
A: The esterification is optimally conducted in polar aprotic solvents like N,N-dimethylformamide (DMF) or acetone, using alkali metal carbonates or bicarbonates as bases. The reaction temperature is typically maintained between 20°C and 80°C, allowing for completion within a short timeframe of 15 minutes to 5 hours, which enhances throughput.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the patent explicitly highlights the method's suitability for industrial implementation due to its operational simplicity and mild reaction conditions. The use of common reagents and the avoidance of complex transition metal catalysts make it highly scalable and economically viable for commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Olmesartan Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from a patented laboratory method to a commercial reality requires more than just chemical knowledge; it demands engineering excellence and rigorous quality assurance. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this novel Olmesartan synthesis are fully realized in practice. We maintain stringent purity specifications across all our batches, supported by rigorous QC labs equipped with state-of-the-art analytical instrumentation to detect even trace levels of impurities. Our commitment to quality ensures that every kilogram of Olmesartan intermediate we supply meets the exacting standards required for global regulatory filings.
We invite you to explore how our optimized manufacturing capabilities can enhance your supply chain resilience and cost structure. By leveraging our expertise in process intensification and impurity control, we can help you achieve a sustainable competitive advantage in the ARB market. Please contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our production of high-purity Olmesartan intermediates can serve as a cornerstone for your pharmaceutical development projects.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →