Scalable Stereoselective Synthesis of Falcarinol for Pharmaceutical Applications
Scalable Stereoselective Synthesis of Falcarinol for Pharmaceutical Applications
The pharmaceutical and fine chemical industries are constantly seeking robust, scalable routes for bioactive natural products that traditionally suffer from supply chain bottlenecks due to low natural abundance. A pivotal advancement in this domain is detailed in patent CN102311314A, which discloses a highly efficient stereoselective total synthetic method for Falcarinol, a potent polyacetylene compound found in plants like ginseng and carrots. This technology addresses the critical limitations of previous methods by utilizing a convergent strategy that significantly shortens the synthetic sequence while maintaining high stereochemical integrity. For R&D directors and procurement specialists, this represents a transformative opportunity to secure a reliable supply of this valuable API intermediate without relying on unpredictable agricultural extraction processes.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the acquisition of Falcarinol and related polyacetylenes has been plagued by significant inefficiencies inherent to natural product isolation. These compounds exist in minute quantities within plant matrices, necessitating the processing of massive amounts of biomass to obtain negligible yields, which is economically and environmentally unsustainable. Furthermore, existing synthetic literature describes routes that are excessively linear, often requiring upwards of ten steps with cumulative yields that plummet to impractical levels. Many traditional approaches rely on chiral pool starting materials like D-xylose or D-gluconolactone, which introduce unnecessary complexity and cost due to the extensive protection and deprotection sequences required to manipulate the carbohydrate backbone. These factors collectively create a fragile supply chain vulnerable to raw material price volatility and batch-to-batch inconsistency.
The Novel Approach
The methodology outlined in the referenced patent revolutionizes this landscape by employing a convergent synthesis strategy that divides the target molecule into two manageable fragments: a chiral head group and a lipophilic tail. By initiating the synthesis from commodity chemicals such as acrolein and propargyl alcohol, the process bypasses the need for expensive chiral pool sugars. The route is characterized by its brevity and operational simplicity, utilizing robust reactions like nucleophilic substitution, Lindlar reduction, and the Cadiot-Chodkiewcz coupling. This strategic disconnection not only reduces the overall step count but also allows for the independent optimization of each fragment, thereby enhancing the overall process reliability and facilitating easier purification of intermediates before the final coupling step.
Mechanistic Insights into Asymmetric Addition and Alkyne Coupling
The cornerstone of this synthesis lies in the precise construction of the chiral center at the C-3 position, which is critical for the biological activity of Falcarinol. This is achieved through an asymmetric addition reaction where diethyl zinc reacts with acrolein in the presence of a chiral ligand, specifically BINOL (1,1'-bi-2-naphthol), and a Lewis acid catalyst like tetraisopropoxy titanium. The choice of the BINOL enantiomer dictates the stereochemical outcome; using (R)-BINOL yields the (S)-configured alcohol, while (S)-BINOL leads to the (R)-isomer. This catalytic system creates a rigid chiral environment that directs the nucleophilic attack of the organozinc species to one face of the aldehyde, ensuring high enantiomeric excess. Following this, the hydroxyl group is preserved while the terminal alkyne is functionalized into a bromo-alkyne, a crucial electrophile for the subsequent coupling.
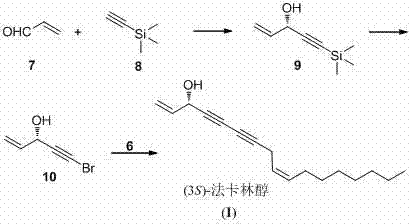
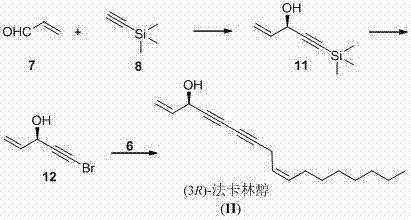
The second critical mechanistic phase involves the assembly of the carbon skeleton via the Cadiot-Chodkiewcz reaction. This cross-coupling reaction joins the terminal alkyne of the lipophilic tail (cis-4-decene-1-yne) with the bromo-alkyne of the chiral head fragment. The reaction is catalyzed by copper salts, such as cuprous chloride or bromide, in the presence of an amine base and hydroxylamine hydrochloride as an oxygen scavenger to prevent oxidative homocoupling (Glaser coupling). The mechanism proceeds through the formation of a copper acetylide species which then undergoes oxidative addition with the bromo-alkyne, followed by reductive elimination to form the conjugated diyne system. This step is remarkably chemoselective, tolerating the cis-alkene geometry established earlier via Lindlar reduction, thus preserving the Z-configuration essential for the native structure of Falcarinol.
How to Synthesize Falcarinol Efficiently
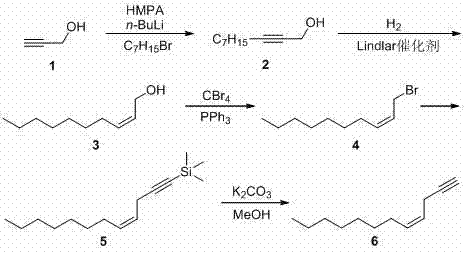
The execution of this synthetic pathway requires careful control of reaction parameters, particularly temperature and stoichiometry, to maximize yield and purity. The process begins with the preparation of the alkyne tail, where propargyl alcohol is alkylated and subsequently reduced to the cis-alkene using a poisoned palladium catalyst to prevent over-reduction to the alkane. Parallel to this, the chiral head fragment is generated under inert atmosphere to protect the sensitive organometallic intermediates. The final coupling is performed at controlled low temperatures to minimize side reactions. For a comprehensive breakdown of the specific reagents, molar ratios, and workup procedures required to implement this chemistry in a pilot or production setting, please refer to the standardized protocol below.
- Synthesize the cis-4-decene-1-yne fragment starting from propargyl alcohol via alkylation, Lindlar reduction, bromination, and desilylation.
- Prepare the chiral 5-bromo-1-pentene-4-yne-3-alcohol fragment via asymmetric addition of ethynyl zinc to acrolein using BINOL/Ti catalysts, followed by bromination.
- Perform the final Cadiot-Chodkiewcz coupling reaction between the two fragments using copper catalysis to yield (3R)- or (3S)-Falcarinol.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this synthetic route offers profound advantages that directly address the pain points of sourcing complex natural product intermediates. By shifting production from agriculture-dependent extraction to chemical synthesis, manufacturers can decouple supply from seasonal harvest variations and geopolitical instability affecting botanical raw materials. The use of bulk commodity chemicals as starting materials ensures a stable and predictable cost base, shielding the supply chain from the volatility associated with niche natural extracts. Furthermore, the high yields reported in the patent embodiments suggest a process that is materially efficient, reducing the volume of waste generated per kilogram of product and lowering the overall cost of goods sold.
- Cost Reduction in Manufacturing: The elimination of expensive chiral pool starting materials like sugars drastically lowers the raw material input costs. Additionally, the shortness of the synthetic route reduces the consumption of solvents, reagents, and energy associated with multiple isolation and purification steps. The high atom economy of the coupling reactions further contributes to substantial cost savings, making the synthetic Falcarinol economically competitive with, if not superior to, extracted material.
- Enhanced Supply Chain Reliability: Chemical synthesis provides a consistent, year-round production capability that is immune to the vagaries of crop yields or climate change impacts on plant growth. The robustness of the reaction conditions, which do not require cryogenic temperatures or ultra-high pressures, simplifies the engineering requirements for manufacturing facilities. This reliability ensures that downstream pharmaceutical customers can maintain continuous production schedules without the risk of raw material shortages.
- Scalability and Environmental Compliance: The process utilizes standard unit operations and common solvents, facilitating straightforward scale-up from laboratory to multi-ton production. The avoidance of heavy metal catalysts in the final steps, relying instead on copper which is easier to manage and remove, simplifies the purification process and aids in meeting stringent regulatory limits for residual metals in API intermediates. This aligns with modern green chemistry principles by minimizing waste and improving the overall environmental footprint of the manufacturing process.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of Falcarinol synthesized via this method. These insights are derived directly from the technical specifications and experimental data provided in the patent documentation, offering clarity on the feasibility and quality of the resulting product. Understanding these details is crucial for technical teams evaluating the integration of this intermediate into their own drug development pipelines.
Q: What are the key advantages of this synthetic route compared to extraction?
A: Unlike plant extraction which yields very low quantities and requires complex purification, this synthetic method offers high yields per step, mild reaction conditions, and excellent stereoselectivity, ensuring a consistent supply of high-purity material.
Q: How is stereocontrol achieved in the synthesis of Falcarinol?
A: Stereocontrol is achieved during the formation of the chiral propargylic alcohol intermediate. By using specific enantiomers of BINOL (either R or S) in conjunction with tetraisopropoxy titanium and diethyl zinc, the process selectively produces either the (3R) or (3S) configuration with high optical purity.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the route utilizes readily available raw materials like acrolein and propargyl alcohol and avoids extremely harsh conditions. The steps involve standard unit operations such as extraction and column chromatography, making it highly amenable to scale-up for industrial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Falcarinol Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic value of advanced synthetic methodologies like the one described in CN102311314A for securing the supply of high-value bioactive compounds. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from bench-scale discovery to industrial manufacturing is seamless and efficient. We are committed to delivering Falcarinol and related polyacetylene intermediates with stringent purity specifications, supported by our rigorous QC labs that utilize state-of-the-art analytical instrumentation to verify identity and potency.
We invite procurement leaders and R&D directors to engage with us for a Customized Cost-Saving Analysis tailored to your specific volume requirements. By leveraging our optimized process capabilities, we can help you reduce lead time for high-purity pharmaceutical intermediates while ensuring full regulatory compliance. Please contact our technical procurement team today to request specific COA data and route feasibility assessments for your upcoming projects.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →