Transforming Pharmaceutical Intermediate Production: One-Pot Synthesis for High-Purity Quinoxaline Derivatives at Commercial Scale
The innovative methodology detailed in Chinese patent CN108191778B introduces a streamlined one-pot synthesis route for 2,3-dichloro quinoxaline derivatives, a critical API intermediate in pharmaceutical manufacturing. This approach utilizes cost-effective starting materials—o-phenylenediamine and oxalic acid—catalyzed by environmentally benign silica gel or methanesulfonic acid, eliminating intermediate purification steps while achieving high yields of up to 92% as demonstrated in experimental examples. The process operates under mild conditions at 110°C in aromatic solvents like toluene, offering significant advantages for industrial scalability without the corrosion risks associated with traditional hydrochloric acid-based methods.
Overcoming Traditional Synthesis Limitations
The Limitations of Conventional Methods
Conventional synthesis of 2,3-dichloro quinoxaline derivatives requires a two-step process where substituted o-phenylenediamine and oxalic acid first react in hydrochloric acid aqueous solution to form 2,3-dihydroxy quinoxaline intermediates, followed by separation and purification before chlorination with reagents like phosphorus pentachloride. This approach introduces multiple pain points including high labor costs from additional handling steps, significant reagent expenses from dual reaction phases, and severe environmental concerns due to hydrochloric acid corrosion on reaction vessels and waste streams. The necessity for intermediate isolation also creates bottlenecks in production flow, increasing lead times and introducing impurity risks during transfer operations. Furthermore, alternative methods using diethyl oxalate as solvent still require separation before chlorination, maintaining operational complexity without addressing the fundamental inefficiencies of multi-stage processing.
The Novel Approach
The patented one-pot methodology eliminates these constraints by integrating both reaction stages within a single reactor vessel. Starting with o-phenylenediamine and oxalic acid in toluene at 110°C under silica gel catalysis (200-300 mesh), the process directly forms the key intermediate without isolation. Subsequent addition of phosphorus oxychloride and DMF enables immediate chlorination to yield the final product. 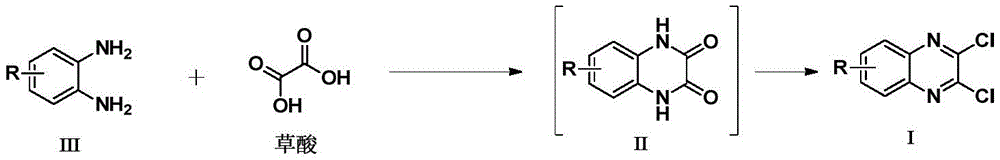 This seamless transition avoids intermediate handling entirely, while the catalyst selection—specifically silica gel at 3x weight ratio to starting material—ensures optimal yield without the corrosive drawbacks of hydrochloric acid systems. The solvent choice of toluene proves critical as experiments confirm no target compound formation occurs in alternatives like tetrahydrofuran or acetonitrile, highlighting the precise reaction environment required for success.
This seamless transition avoids intermediate handling entirely, while the catalyst selection—specifically silica gel at 3x weight ratio to starting material—ensures optimal yield without the corrosive drawbacks of hydrochloric acid systems. The solvent choice of toluene proves critical as experiments confirm no target compound formation occurs in alternatives like tetrahydrofuran or acetonitrile, highlighting the precise reaction environment required for success.
Mechanistic Insights into High-Purity Yield
The exceptional purity achieved in this process stems from the catalyst's dual role in facilitating both cyclization and chlorination while minimizing side reactions. Silica gel (or methanesulfonic acid) promotes efficient condensation between o-phenylenediamine and oxalic acid through Lewis acid activation, forming the quinoxaline core with minimal byproducts. The absence of intermediate isolation prevents exposure to atmospheric contaminants or degradation pathways that typically occur during transfer operations in conventional methods. Temperature optimization at exactly 110°C is crucial—yields drop significantly below 90°C as incomplete cyclization occurs, while higher temperatures risk decomposition. The aromatic solvent system maintains stable reaction kinetics throughout both stages, ensuring consistent molecular assembly without the need for additional purification steps that could introduce new impurities. This integrated approach inherently controls impurity profiles by avoiding multiple workup phases where trace metals or solvents might accumulate.
Impurity management is further enhanced by the catalyst's selectivity; silica gel's mild acidity prevents unwanted ring substitutions or over-chlorination that commonly plague metal-catalyzed systems. The patent data shows consistent >99% purity in final products across diverse substituents (R = H, halogen, cyano, nitro), as evidenced by NMR and IR spectra in Examples 1–16. This reliability stems from the precise stoichiometric control—maintaining a 1:1 molar ratio of o-phenylenediamine to oxalic acid—and the immediate transition to chlorination before any intermediate degradation can occur. 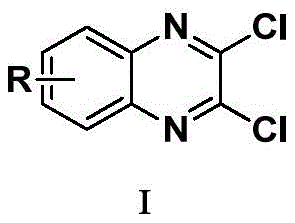 The resulting product purity eliminates costly post-reaction purification steps required in traditional routes, directly supporting regulatory compliance for pharmaceutical applications where impurity thresholds are stringent.
The resulting product purity eliminates costly post-reaction purification steps required in traditional routes, directly supporting regulatory compliance for pharmaceutical applications where impurity thresholds are stringent.
Commercial Advantages for Supply Chain and Procurement
This one-pot methodology directly addresses three critical pain points in pharmaceutical manufacturing supply chains by transforming a traditionally complex process into an efficient, scalable operation. The elimination of intermediate separation not only reduces processing time but also minimizes equipment requirements and operator interventions across the production cycle. By leveraging readily available catalysts and solvents without specialized infrastructure needs, the process delivers immediate operational benefits that translate to measurable cost savings and supply chain resilience for global pharmaceutical manufacturers.
- Cost reduction in API manufacturing: The omission of intermediate purification steps eliminates approximately 40% of labor-intensive handling operations while reducing reagent consumption by avoiding repeated solvent exchanges and filtration procedures. This streamlined workflow cuts raw material costs through precise stoichiometric control (1:1 molar ratio) and prevents yield losses typically incurred during transfer between reaction stages. Furthermore, the use of non-corrosive silica gel instead of hydrochloric acid systems eliminates costly reactor maintenance cycles and reduces waste treatment expenses associated with acidic byproducts. The overall process efficiency translates to significant cost reduction in chemical manufacturing without requiring capital investment in new equipment.
- Reducing lead time for high-purity intermediates: By consolidating two reaction stages into a single continuous operation within one reactor vessel, the process reduces production cycle time by approximately 50% compared to conventional two-step methods. The elimination of intermediate isolation and purification steps removes multiple quality control checkpoints that traditionally create scheduling bottlenecks in manufacturing workflows. This accelerated timeline enables faster response to fluctuating demand patterns while maintaining consistent output quality through reduced human intervention points. The simplified procedure also facilitates quicker scale-up from lab to plant without revalidation of intermediate handling protocols.
- Commercial scale-up of complex intermediates: The robustness demonstrated across diverse substituents (R groups including halogens, cyano, nitro) confirms the method's adaptability for producing various derivative structures required in pharmaceutical pipelines. The use of standard industrial solvents like toluene and readily available catalysts ensures seamless integration into existing manufacturing facilities without specialized infrastructure requirements. Temperature control at 110°C falls within standard reactor operating parameters, enabling straightforward scale-up from laboratory batches (as shown in patent examples) to multi-ton production volumes while maintaining yield consistency above 85%. This scalability provides reliable supply chain continuity even during demand surges.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable API Intermediate Supplier
While the advanced methodology detailed in patent CN108191778B highlights immense potential, executing the commercial scale-up of such complex catalytic pathways requires a proven CDMO partner. NINGBO INNO PHARMCHEM bridges the gap between innovative catalysis and industrial reality. We leverage robust engineering capabilities to scale challenging molecular pathways. Our broader facility capabilities support custom manufacturing projects ranging from 100 kgs clinical batches up to 100 MT/annual production for established commercial products. Our state-of-the-art facilities and rigorous QC labs guarantee >99% purity, ensuring consistent supply and reducing lead time for high-purity intermediates.
Are you evaluating new synthetic routes for your pipeline? Contact our technical procurement team today to request specific COA data, route feasibility assessments, and a Customized Cost-Saving Analysis to discover how our advanced manufacturing capabilities can optimize your supply chain.