Revolutionizing Feloxicib Production: A Green, Scalable Synthetic Route for Global Pharma Supply Chains
Revolutionizing Feloxicib Production: A Green, Scalable Synthetic Route for Global Pharma Supply Chains
The pharmaceutical industry is constantly seeking more efficient, sustainable, and cost-effective pathways for the synthesis of active pharmaceutical ingredients (APIs) and their critical intermediates. A significant breakthrough in this domain is detailed in patent CN107686471B, which outlines a novel synthesis method for Feloxicib, a potent nonsteroidal anti-inflammatory drug (NSAID), and its key intermediates. This technology represents a paradigm shift from traditional, hazardous synthetic routes to a greener, more streamlined process that leverages advanced catalytic systems. By replacing toxic starting materials like thioethers with safer alternatives such as p-bromophenylketone, and utilizing a unique copper-catalyzed sulfinylation followed by an NBS-mediated hydroxylation, this method addresses long-standing challenges in impurity control and environmental compliance. For global procurement and R&D teams, understanding this technological leap is crucial for securing a reliable pharmaceutical intermediates supplier capable of delivering high-purity materials with reduced lead times and enhanced supply chain resilience.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Feloxicib has relied on routes that pose significant operational and environmental hazards, creating bottlenecks for large-scale manufacturing. As illustrated in prior art such as US5981576, the conventional pathway typically initiates with thioanisole (thiobenzene), a compound notorious for its intense, unpleasant odor and high flammability, which presents severe safety risks in industrial settings. Furthermore, these legacy processes often necessitate the use of carbon tetrachloride, a Class 1 solvent with known carcinogenic properties, and require complex phase-transfer catalysis systems involving Aliquat 336. The purification of intermediates in these traditional routes frequently demands silica gel column chromatography, a technique that is notoriously difficult to scale and economically unviable for multi-kilogram or ton-level production. Additionally, alternative methods employing liquid bromine for bromination introduce further handling dangers and environmental burdens, while multi-step oxidation sequences using reagents like MMPP extend reaction times and increase waste generation.
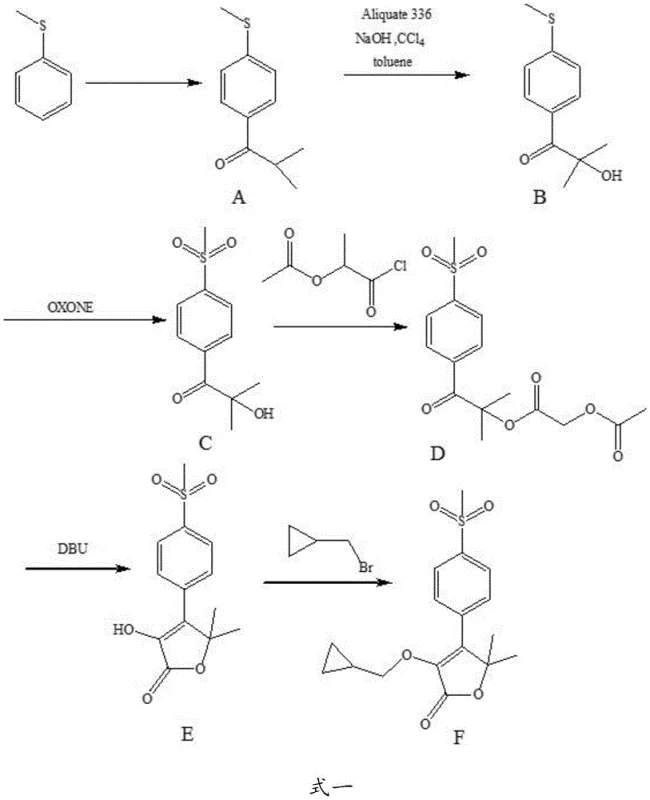
The Novel Approach
In stark contrast, the innovative methodology disclosed in patent CN107686471B fundamentally redesigns the synthetic architecture to prioritize safety and efficiency. This novel approach abandons the problematic thioether starting material in favor of p-bromophenylketone, a stable and commercially accessible solid that eliminates odor and flammability concerns at the source. The core of this advancement lies in a strategic sequence where methylation is followed by a direct sulfinylation reaction catalyzed by cuprous iodide, effectively merging what were previously separate sulfuration and oxidation steps into a single, high-yield transformation. Subsequently, the process employs an N-bromosuccinimide (NBS) system for direct hydroxylation, bypassing the need for hazardous liquid bromine or expensive peroxides. This streamlined workflow not only accelerates the overall reaction rate but also simplifies downstream processing, allowing for purification via simple crystallization rather than chromatography, thereby making the process inherently suitable for commercial scale-up of complex pharmaceutical intermediates.
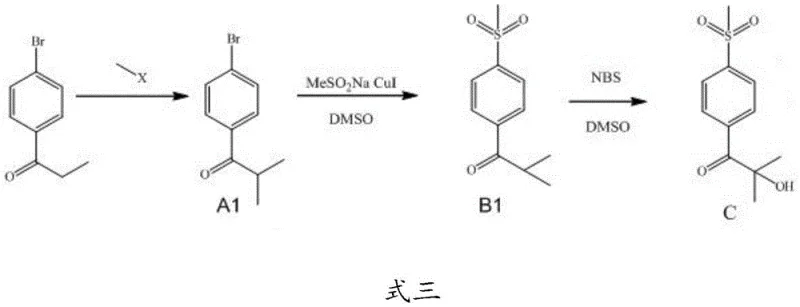
Mechanistic Insights into CuI-Catalyzed Sulfinylation and NBS Oxidation
The mechanistic elegance of this new route is centered on the transition metal-catalyzed introduction of the methylsulfonyl group, a critical structural motif for the biological activity of Feloxicib. In the second step of the synthesis, the first intermediate (1-(4-bromophenyl)-2-methylpropanone) undergoes a nucleophilic substitution where the bromine atom is displaced by a methanesulfinate group. This transformation is facilitated by a catalytic system comprising cuprous iodide (CuI) and L-proline in dimethyl sulfoxide (DMSO) at elevated temperatures (80-120°C). The copper catalyst likely operates through a cycle involving oxidative addition of the aryl bromide to the copper center, followed by transmetallation with the sodium methanesulfinate and subsequent reductive elimination to forge the C-S bond. This one-pot strategy is superior to traditional methods because it installs the sulfur functionality in a higher oxidation state directly, or facilitates rapid subsequent oxidation, thereby collapsing multiple synthetic operations into a single unit operation. The use of L-proline as a ligand enhances the solubility and stability of the copper species, ensuring consistent catalytic turnover and minimizing the formation of homocoupling byproducts that often plague aryl halide substitutions.
Following the installation of the sulfone moiety, the process employs a highly selective hydroxylation mechanism using N-bromosuccinimide (NBS) in DMSO. Unlike traditional alpha-halogenation followed by hydrolysis which can lead to over-halogenation or elimination side reactions, the NBS/DMSO system provides a controlled source of electrophilic bromine that targets the alpha-position of the ketone adjacent to the newly formed sulfone group. The reaction conditions (100°C in DMSO) promote the displacement of the transient alpha-bromo species by oxygen nucleophiles derived from the solvent or trace water, ultimately yielding the tertiary alcohol intermediate with high regioselectivity. This step is critical for impurity control, as the mild nature of NBS compared to elemental bromine reduces the risk of ring bromination or degradation of the sensitive sulfone group. The resulting intermediate precipitates cleanly upon workup, demonstrating the robustness of the chemical design in maintaining product integrity throughout the synthesis.
How to Synthesize Feloxicib Intermediate Efficiently
Implementing this advanced synthesis protocol requires precise control over reaction parameters to maximize yield and purity, particularly during the catalytic sulfinylation and hydroxylation stages. The process begins with the methylation of p-bromophenylketone under basic conditions, followed by the critical copper-catalyzed step which demands strict temperature management between 80°C and 120°C to ensure complete conversion without degrading the ligand system. The final hydroxylation step utilizes a specific molar ratio of NBS to substrate to prevent over-oxidation while driving the reaction to completion. Detailed standard operating procedures regarding solvent ratios, addition rates, and crystallization protocols are essential for reproducibility. For a comprehensive guide on executing this synthesis with optimal results, please refer to the standardized technical instructions below.
- Methylation of p-bromophenylketone using methyl iodide and strong base in DMF or DMSO to form Intermediate A1.
- CuI-catalyzed sulfinylation of Intermediate A1 with sodium methanesulfinate in DMSO at 100°C to yield Intermediate B1.
- Direct hydroxylation of Intermediate B1 using N-bromosuccinimide (NBS) in DMSO to obtain the critical Feloxicib precursor.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthesis route offers tangible strategic benefits that extend beyond mere chemical curiosity. By eliminating the reliance on controlled or hazardous substances like carbon tetrachloride and liquid bromine, manufacturers can significantly reduce the regulatory burden and safety compliance costs associated with storage, handling, and waste disposal. The shift away from thioether starting materials also mitigates the risk of production shutdowns due to odor complaints or safety incidents, ensuring a more stable and continuous supply of critical intermediates. Furthermore, the removal of silica gel chromatography from the purification train translates directly into substantial cost savings, as it reduces solvent consumption, labor hours, and equipment downtime, thereby enhancing the overall economic viability of the manufacturing process.
- Cost Reduction in Manufacturing: The streamlined nature of this synthesis directly impacts the bottom line by consolidating multiple reaction steps into fewer, higher-yielding operations. By avoiding expensive oxidants like MMPP and eliminating the need for resource-intensive column chromatography, the process significantly lowers the cost of goods sold (COGS). The use of common, commercially available reagents such as sodium methanesulfinate and NBS ensures that raw material costs remain stable and predictable, shielding the supply chain from volatility associated with specialty chemicals. Additionally, the high purity achieved through simple crystallization reduces the need for reprocessing, further optimizing resource utilization and driving down the total cost of production for high-purity pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: The choice of p-bromophenylketone as a starting material provides a robust foundation for supply security, as it is a widely produced commodity chemical with multiple global sources, unlike niche thioethers which may have limited suppliers. The avoidance of carcinogenic solvents like carbon tetrachloride removes a major bottleneck in logistics and transportation, as shipping hazardous Class 1 solvents often incurs delays and requires specialized handling infrastructure. This logistical simplicity ensures that production schedules can be maintained without interruption, providing partners with a dependable source of materials and reducing lead time for high-purity pharmaceutical intermediates essential for downstream API synthesis.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this route is designed for scalability, adhering to the principles of green chemistry by minimizing waste and avoiding toxic reagents. The ability to purify intermediates via crystallization rather than chromatography makes the process inherently easier to scale from pilot plant to multi-ton commercial production without losing efficiency. This scalability ensures that the supply chain can flexibly respond to market demand surges for Feloxicib. Moreover, the reduced environmental footprint aligns with the increasingly stringent sustainability goals of multinational pharmaceutical companies, facilitating smoother regulatory approvals and fostering long-term partnerships focused on eco-friendly cost reduction in pharmaceutical intermediates manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent literature, offering clarity on how this method outperforms legacy routes in terms of safety, efficiency, and product quality. Understanding these nuances is vital for technical teams evaluating the feasibility of integrating this pathway into their existing manufacturing portfolios.
Q: Why is the new Feloxicib synthesis route considered more environmentally friendly?
A: The new route eliminates the use of malodorous and flammable thioethers as starting materials and avoids carcinogenic solvents like carbon tetrachloride and hazardous reagents like liquid bromine, significantly reducing environmental impact and safety risks.
Q: How does the CuI-catalyzed step improve production efficiency?
A: By utilizing cuprous iodide and sodium methanesulfinate, the process combines sulfuration and oxidation into a single substitution step, drastically reducing reaction time and simplifying the workflow compared to traditional multi-step oxidation methods.
Q: Is this synthesis method suitable for large-scale industrial manufacturing?
A: Yes, the method avoids column chromatography purification, relies on common commercially available reagents, and utilizes robust crystallization techniques, making it highly adaptable for ton-scale commercial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Feloxicib Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition to advanced synthetic routes requires a partner with deep technical expertise and proven manufacturing capabilities. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of patent CN107686471B are fully realized in practical, industrial applications. We are committed to delivering materials that meet stringent purity specifications through our rigorous QC labs, guaranteeing that every batch of Feloxicib intermediate supports your downstream synthesis with consistency and reliability. Our facility is equipped to handle the specific catalytic and crystallization requirements of this green chemistry route, positioning us as a strategic ally in your supply chain.
We invite you to collaborate with us to leverage these technological advancements for your next project. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our optimized synthesis of Feloxicib intermediates can enhance your operational efficiency and drive value across your organization.