Advanced Synthesis of Paeonol Dihydropyrimidinone Derivatives for Oncology Applications
The pharmaceutical landscape is constantly evolving with the demand for more effective antitumor agents that minimize systemic toxicity. Patent CN110437156B introduces a significant breakthrough in this domain by disclosing a novel class of paeonol dihydropyrimidinone derivatives. These compounds represent a strategic fusion of the natural product paeonol, known for its anti-inflammatory and antitumor properties, with the pharmacologically privileged dihydropyrimidinone scaffold. This hybridization aims to enhance bioavailability and target specificity while reducing adverse side effects. The patent outlines a robust preparation method that leverages modular synthetic steps, ensuring high purity and stable quality of the final derivatives. For research and development teams seeking reliable pharmaceutical intermediate suppliers, this technology offers a promising pathway for developing next-generation oncology therapeutics.
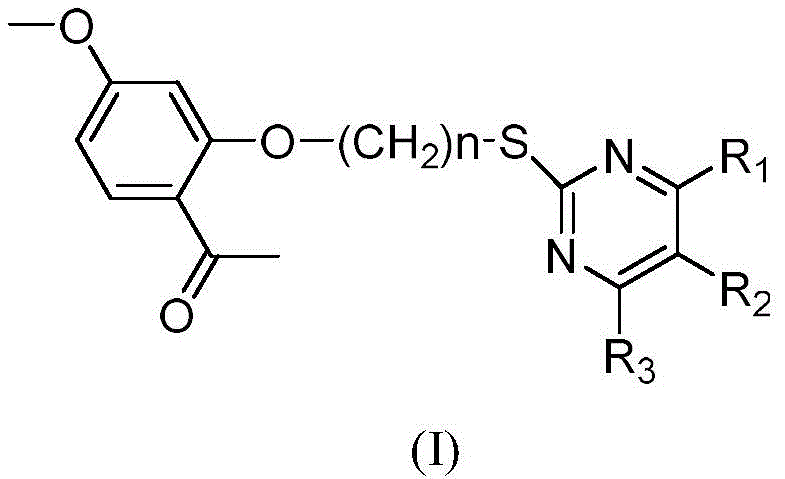
The structural versatility of these derivatives is defined by the general formula (I), where substituents R1, R2, and R3 can be varied to optimize pharmacological profiles. The inclusion of a linker chain, represented by (CH2)n where n ranges from 1 to 6, provides conformational flexibility that is crucial for receptor binding. This molecular architecture is designed to improve the interaction with biological targets compared to the parent paeonol molecule. By systematically modifying the aromatic aldehyde component used in the synthesis, a diverse library of analogs can be generated, allowing for extensive structure-activity relationship (SAR) studies. This approach is critical for identifying lead compounds with superior efficacy against resistant tumor cell lines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional methods for modifying paeonol often involve direct esterification or etherification which may lack the structural complexity required for potent antitumor activity. Many existing synthetic routes rely on harsh reaction conditions, expensive transition metal catalysts, or multi-step protection and deprotection sequences that significantly increase production costs and environmental waste. Furthermore, conventional derivatives often suffer from poor water solubility and rapid metabolic clearance, limiting their therapeutic window. The inability to easily introduce heterocyclic motifs like dihydropyrimidinones using standard protocols has historically restricted the exploration of this chemical space. Consequently, there has been a lack of public reports combining paeonol with dihydropyrimidinone moieties until the innovations presented in this patent.
The Novel Approach
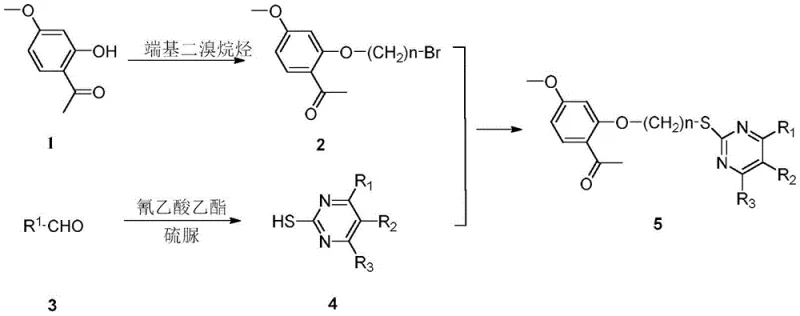
The novel approach detailed in the patent overcomes these hurdles through a streamlined three-step synthesis that avoids complex catalytic systems. The strategy employs a nucleophilic substitution reaction to attach a bromoalkyl chain to the paeonol core, followed by a separate cyclization to form the dihydropyrimidinone ring. Finally, these two fragments are coupled via another nucleophilic substitution. This convergent synthesis allows for the independent optimization of each fragment before assembly, greatly enhancing the overall efficiency. The use of mild bases like anhydrous potassium carbonate and common solvents such as ethanol ensures that the process is both economically viable and environmentally friendlier. This methodology facilitates the commercial scale-up of complex pharmaceutical intermediates by simplifying purification and reducing energy consumption.
Mechanistic Insights into Nucleophilic Substitution and Cyclization
The core of this synthesis relies on precise control of nucleophilic substitution mechanisms. In the first step, the phenolic hydroxyl group of paeonol acts as a nucleophile, attacking the terminal carbon of a dibromoalkane. The presence of an acid-binding agent, preferably anhydrous potassium carbonate, is essential to deprotonate the phenol, generating a phenoxide ion that is significantly more reactive. This reaction is typically conducted at temperatures between 30°C and 80°C, which is mild enough to prevent degradation of the sensitive acetophenone moiety while providing sufficient energy for the substitution to proceed. The molar ratio of paeonol to the dibromoalkane is carefully controlled, often using an excess of the alkylating agent to drive the reaction to completion and minimize dialkylation byproducts.
The formation of the dihydropyrimidinone ring involves a cyclocondensation reaction reminiscent of the Biginelli reaction but adapted for thiourea derivatives. Aromatic aldehydes react with ethyl cyanoacetate and thiourea in the presence of a catalyst to form the 6-substituted phenyl-5-cyano-2-thiouracil intermediate. This step creates the heterocyclic core that is responsible for much of the biological activity. The final coupling step involves the nucleophilic attack of the sulfur atom in the thiouracil intermediate on the bromo-paeonol fragment. This thioether linkage is stable and serves as an effective bridge between the two pharmacophores. Understanding these mechanistic details is vital for cost reduction in pharmaceutical intermediate manufacturing, as it allows for the minimization of side reactions and maximizes atom economy.
How to Synthesize Paeonol Dihydropyrimidinone Derivatives Efficiently
The synthesis protocol described in the patent provides a clear roadmap for producing these high-value compounds. The process begins with the preparation of the brominated paeonol intermediate, followed by the synthesis of the thiouracil derivative, and concludes with the coupling reaction. Each step is monitored using thin-layer chromatography (TLC) to ensure reaction completeness before proceeding to purification. The purification typically involves silica gel column chromatography, yielding products with high purity suitable for biological testing. For detailed operational parameters and specific stoichiometric ratios, please refer to the standardized guide below.
- Perform nucleophilic substitution on paeonol with a terminal dibromoalkane in an organic solvent with an acid-binding agent to generate brominated paeonol.
- Conduct a cyclization reaction using aromatic aldehyde, ethyl cyanoacetate, and thiourea in the presence of a catalyst to synthesize 6-substituted phenyl-5-cyano-2-thiouracil.
- Execute a final nucleophilic substitution between the brominated paeonol and the synthesized thiouracil derivative in an organic solvent to obtain the target paeonol dihydropyrimidinone derivative.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, the synthetic route outlined in patent CN110437156B offers substantial advantages in terms of raw material availability and process safety. The starting materials, including paeonol, aromatic aldehydes, and thiourea, are commodity chemicals that are readily available from multiple global suppliers, mitigating supply chain risks associated with single-source dependencies. The avoidance of precious metal catalysts eliminates the need for expensive metal scavenging steps and reduces the burden of heavy metal residue testing in the final API. This translates directly into lower production costs and faster time-to-market for new drug candidates. Additionally, the use of common solvents like ethanol simplifies solvent recovery and recycling processes, further enhancing the economic viability of the manufacture.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts and the use of mild reaction conditions significantly lower the operational expenditure. By avoiding high-pressure or high-temperature requirements, the process reduces energy consumption and equipment wear. The high selectivity of the nucleophilic substitution steps minimizes the formation of difficult-to-remove impurities, thereby reducing the cost of downstream purification. This efficiency is crucial for maintaining competitive pricing in the generic drug market while ensuring high margins for innovative therapies.
- Enhanced Supply Chain Reliability: The modular nature of the synthesis allows for the stockpiling of key intermediates, such as the brominated paeonol and various thiouracil derivatives. This flexibility enables manufacturers to respond quickly to fluctuations in demand without disrupting the entire production line. Since the reaction conditions are not sensitive to trace moisture or oxygen to an extreme degree, the process is robust and less prone to batch failures. This reliability ensures a consistent supply of high-quality intermediates, which is a critical factor for pharmaceutical companies managing tight production schedules.
- Scalability and Environmental Compliance: The process is inherently scalable from gram to kilogram and eventually to tonnage levels without significant re-engineering. The solvents used are generally less toxic and easier to handle than chlorinated alternatives often found in older methodologies. Furthermore, the byproducts of the reaction are primarily inorganic salts which can be treated using standard wastewater management protocols. This alignment with green chemistry principles helps companies meet increasingly stringent environmental regulations and sustainability goals, avoiding potential fines and reputational damage.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these derivatives. The answers are derived directly from the experimental data and claims within the patent documentation, providing accurate guidance for potential partners and licensees. Understanding these details is essential for evaluating the feasibility of integrating this technology into existing R&D pipelines.
Q: What are the key structural features of the paeonol dihydropyrimidinone derivatives described in patent CN110437156B?
A: The derivatives feature a paeonol skeleton linked via an alkyl chain to a dihydropyrimidinone ring. The structure allows for variability at the R1 position (aryl, H, or CH3), R2 (CN or H), and R3 (OH or CH3), enabling fine-tuning of biological activity.
Q: How does the antitumor activity of these derivatives compare to standard treatments?
A: Specific derivatives, such as compound 5i, have demonstrated superior inhibitory activity against HCT-116 colon cancer cells compared to cisplatin, with significantly lower toxicity towards normal liver cells (LO2).
Q: Is the synthesis process scalable for industrial production?
A: Yes, the process utilizes common organic solvents like ethanol and acetonitrile, mild reaction temperatures ranging from 30°C to 80°C, and readily available catalysts such as potassium carbonate, making it highly suitable for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Paeonol Dihydropyrimidinone Derivatives Supplier
NINGBO INNO PHARMCHEM stands at the forefront of custom synthesis and contract development, possessing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the specific requirements of synthesizing paeonol derivatives, ensuring stringent purity specifications and rigorous QC labs verify every batch. We understand the critical nature of oncology intermediates and are committed to delivering materials that meet the highest international standards for clinical and commercial use. Our team of expert chemists is ready to assist in optimizing the synthetic route for your specific needs, ensuring maximum yield and minimal impurity profiles.
We invite you to contact our technical procurement team to discuss your project requirements in detail. By partnering with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume and purity needs. We encourage potential clients to request specific COA data and route feasibility assessments to validate our capabilities. Let us help you accelerate your drug development timeline with our reliable supply chain and technical expertise in complex heterocyclic synthesis.