Scalable Metal-Free Synthesis of Imidazole Antiviral Compounds for Global Pharma Supply Chains
Scalable Metal-Free Synthesis of Imidazole Antiviral Compounds for Global Pharma Supply Chains
The pharmaceutical industry is constantly seeking robust, scalable, and cost-effective pathways for the production of active pharmaceutical ingredients (APIs) and their critical intermediates. A significant breakthrough in this domain is documented in Chinese Patent CN111303039A, which details a novel preparation method for a specific imidazole antiviral compound. This technology represents a paradigm shift from traditional transition-metal catalyzed processes to a more sustainable and economically viable organocatalytic approach. By leveraging a unique tandem cyclization strategy followed by streamlined functional group transformations, this method addresses key pain points in antiviral drug manufacturing, specifically regarding catalyst cost, step count, and overall yield. For R&D directors and procurement managers alike, understanding the nuances of this four-step sequence is crucial for optimizing supply chains and ensuring the consistent availability of high-purity antiviral intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art in the synthesis of imidazole-based antiviral agents, such as the methodology described in WO 2018178167, has historically relied on multi-step sequences that often necessitate the use of expensive transition metal catalysts. These conventional routes typically involve five or more distinct chemical transformations, each introducing potential points of failure, yield loss, and impurity generation. The reliance on precious metals not only inflates the raw material costs but also imposes stringent regulatory burdens regarding residual metal limits in the final drug substance. Furthermore, longer synthetic linearities inherently increase the lead time for production and complicate the supply chain logistics, making it difficult to respond rapidly to market demands for antiviral therapies. The accumulation of impurities over multiple steps often requires complex chromatographic purifications, which are notoriously difficult to scale and significantly drive up the cost of goods sold (COGS).
The Novel Approach
In stark contrast, the methodology outlined in CN111303039A introduces a streamlined four-step synthetic route that elegantly bypasses the need for expensive metal catalysts. The core innovation lies in the initial tandem cyclization reaction, which constructs the complex biaryl scaffold in a single operational step using an organocatalytic system. This reduction in step count from five to four may seem marginal on paper, but in industrial practice, it translates to substantial improvements in overall throughput and material efficiency. By eliminating the metal-catalyzed step, the process removes the need for costly scavenging resins and extensive metal testing, thereby simplifying the downstream processing. The use of readily available reagents such as pyridine p-toluenesulfonate and diphenylmethylphosphine ensures that the supply chain remains resilient against fluctuations in the pricing of rare earth elements or precious metals, offering a distinct competitive advantage in cost reduction in pharmaceutical intermediates manufacturing.
Mechanistic Insights into PPTS/Ph2PMe Co-Catalyzed Tandem Cyclization
The heart of this synthetic innovation is the first step: a tandem cyclization between 1,3-di(2,4-dichloro)phenyl-2-propyn-1-ol and methyl allenoate. This transformation is driven by a synergistic co-catalytic system comprising pyridine p-toluenesulfonate (PPTS) and diphenylmethylphosphine (Ph2PMe). Mechanistically, the phosphine acts as a nucleophilic catalyst, attacking the electron-deficient allenoate to generate a zwitterionic intermediate. Simultaneously, the mild Brønsted acid PPTS activates the propargylic alcohol, facilitating the departure of the hydroxyl group or enhancing the electrophilicity of the alkyne moiety. This dual activation allows for a cascade of bond-forming events that construct the central benzene ring and establish the critical biaryl linkage in one pot. The reaction proceeds efficiently in toluene under heating conditions, typically around 50°C to 70°C, demonstrating remarkable chemoselectivity and tolerance for the chloro-substituents on the aromatic rings.
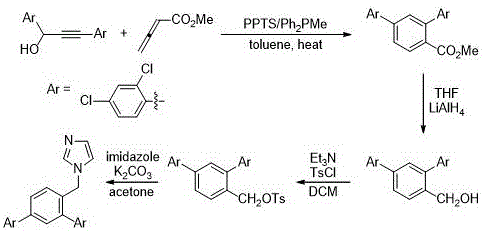
Following the construction of the core scaffold, the subsequent steps are designed for maximum robustness and scalability. The resulting methyl 2,4-diarylbenzoate is subjected to reduction using lithium aluminum hydride (LiAlH4) in tetrahydrofuran (THF). While LiAlH4 is a potent reducing agent, its use here is justified by the need to quantitatively convert the ester to the primary alcohol without affecting the sensitive chloro-substituents or the newly formed aromatic system. The resulting 2,4-diarylbenzyl alcohol is then activated via tosylation using p-toluenesulfonyl chloride (TsCl) and triethylamine. This conversion of the hydroxyl group into a tosylate is a strategic move; the tosylate is an excellent leaving group, far superior to the hydroxyl group itself, which primes the molecule for the final nucleophilic substitution. In the final step, imidazole acts as the nucleophile, displacing the tosylate in the presence of potassium carbonate in acetone. This SN2-type displacement installs the pharmacophore imidazole ring, completing the synthesis of the target antiviral compound with high fidelity.
How to Synthesize Imidazole Antiviral Compound Efficiently
Executing this synthesis requires careful attention to stoichiometry and reaction conditions to maximize the impressive yields reported in the patent data. The process begins with the precise mixing of the propargylic alcohol and allenoate in a molar ratio ranging from 1:1 to 1:1.3, ensuring complete consumption of the limiting reagent. The catalyst loading is optimized between 5 mol% and 20 mol%, balancing reaction rate with cost efficiency. Following the cyclization, the workup involves a standard aqueous extraction and crystallization from n-hexane, which effectively removes polar impurities and catalyst residues. The subsequent reduction and tosylation steps are performed under controlled temperatures, often utilizing ice-water baths during reagent addition to manage exotherms, followed by ambient stirring to ensure completion. For the detailed standardized operating procedures, including specific quenching protocols and isolation techniques, please refer to the technical guide below.
- Perform a tandem cyclization of 1,3-di(2,4-dichloro)phenyl-2-propyn-1-ol with methyl allenoate using PPTS and Ph2PMe catalysts in toluene.
- Reduce the resulting methyl 2,4-diarylbenzoate intermediate to the corresponding benzyl alcohol using lithium aluminum hydride in THF.
- Convert the hydroxyl group into a leaving group by reacting with p-toluenesulfonyl chloride and triethylamine in dichloromethane.
- Complete the synthesis via nucleophilic substitution with imidazole and potassium carbonate in acetone under reflux.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers compelling economic and logistical benefits that extend beyond simple yield metrics. The most significant advantage is the drastic simplification of the raw material portfolio. By replacing expensive transition metal catalysts with commodity organic chemicals like PPTS and phosphines, the direct material costs are significantly reduced. Furthermore, the elimination of heavy metals removes a major bottleneck in quality control; there is no longer a need for specialized ICP-MS testing for residual palladium or other metals, nor the procurement of expensive metal scavengers, leading to substantial cost savings in the analytical and purification phases of manufacturing.
- Cost Reduction in Manufacturing: The economic impact of this metal-free protocol is profound. Transition metal catalysts are not only expensive to purchase but often require specialized ligands and inert atmosphere handling, which increases operational expenditures. By shifting to an organocatalytic system that operates in common solvents like toluene and acetone, the process becomes compatible with standard glass-lined or stainless steel reactors without the need for exotic metallurgy. The high conversion rates observed in the tandem cyclization step minimize the formation of difficult-to-separate byproducts, thereby increasing the overall mass efficiency of the plant. This efficiency translates directly into a lower cost per kilogram of the final API intermediate, providing a stronger margin profile for the finished drug product.
- Enhanced Supply Chain Reliability: Supply chain resilience is heavily dependent on the availability of starting materials. The reagents utilized in this four-step sequence, including methyl allenoate, imidazole, and tosyl chloride, are bulk commodities produced by numerous global suppliers. This diversification of the supply base mitigates the risk of shortages that often plague specialized catalytic reagents. Additionally, the robustness of the chemistry means that the process is less sensitive to minor variations in reagent quality, reducing the likelihood of batch failures. This reliability ensures consistent delivery schedules to downstream API manufacturers, preventing production delays that could impact patient access to critical antiviral medications.
- Scalability and Environmental Compliance: From an environmental and safety perspective, this route aligns well with green chemistry principles. The reduction in step count inherently reduces the total solvent consumption and waste generation per unit of product. The avoidance of toxic heavy metals simplifies waste stream management and disposal, lowering the environmental compliance burden on the manufacturing facility. The reactions utilize standard workup procedures such as liquid-liquid extraction and crystallization, which are easily scalable from pilot plant to commercial multi-ton production. The high yields achieved in each step, particularly the near-quantitative tosylation and substitution, ensure that the process remains efficient even at large scales, supporting the commercial scale-up of complex pharmaceutical intermediates without compromising on purity or safety standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent literature, offering a clear picture of the process capabilities. Understanding these details is essential for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: What are the primary advantages of this metal-free synthesis route?
A: The primary advantages include the elimination of expensive transition metal catalysts, which simplifies purification and reduces heavy metal residue risks. Additionally, the process shortens the synthetic sequence to four steps with a total yield exceeding 78%, significantly improving cost-efficiency compared to prior five-step metal-catalyzed methods.
Q: How does the PPTS/Ph2PMe co-catalytic system improve reaction efficiency?
A: The co-catalytic system enables a highly efficient tandem cyclization between the propargylic alcohol and the allenoate. Pyridine p-toluenesulfonate activates the substrate while diphenylmethylphosphine facilitates the cyclization, allowing the reaction to proceed under mild heating in toluene with high conversion rates and minimal byproduct formation.
Q: Is this synthetic route suitable for large-scale commercial production?
A: Yes, the route is highly suitable for scale-up. It utilizes widely available commercial reagents, avoids sensitive metal catalysts that require specialized handling, and employs standard workup procedures like extraction and crystallization, ensuring robust supply chain continuity and ease of manufacturing at multi-ton scales.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Imidazole Antiviral Compound Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient, scalable, and compliant synthesis routes in the modern pharmaceutical landscape. Our team of expert process chemists has thoroughly analyzed the technology presented in CN111303039A and is fully equipped to translate this laboratory-scale innovation into commercial reality. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. Our state-of-the-art facilities are designed to handle complex organic syntheses, including those requiring stringent purity specifications and rigorous QC labs to guarantee that every batch meets the highest international standards for antiviral intermediates.
We invite you to collaborate with us to leverage this advanced metal-free technology for your antiviral drug development programs. By partnering with our technical procurement team, you can gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to reach out today to request specific COA data and route feasibility assessments, allowing us to demonstrate how our manufacturing expertise can optimize your supply chain and accelerate your time to market.