Advanced Catalytic Synthesis of Chiral Acyclic Nucleosides for Commercial API Manufacturing
Introduction to Next-Generation Nucleoside Synthesis
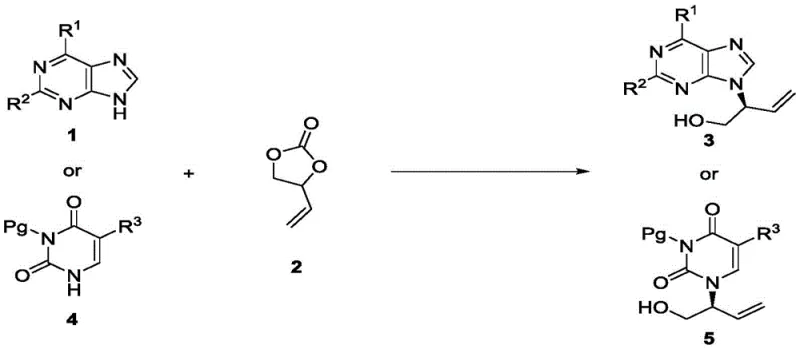
The synthesis of chiral acyclic nucleosides represents a critical frontier in the development of antiviral therapeutics, serving as the structural backbone for blockbuster drugs such as Acyclovir, Ganciclovir, and Tenofovir. Historically, accessing these high-value intermediates has been fraught with synthetic challenges, often requiring cumbersome multi-step sequences that erode overall yield and escalate production costs. However, a significant technological breakthrough documented in patent CN112759595B introduces a transformative approach utilizing palladium-catalyzed asymmetric allylation. This methodology bypasses the traditional reliance on expensive chiral pools, instead constructing the stereocenter directly from achiral precursors with exceptional precision. For pharmaceutical manufacturers and procurement strategists, this innovation signals a paradigm shift towards more sustainable and economically viable supply chains for complex nucleoside analogs.
The core of this invention lies in the direct coupling of substituted purines or pyrimidines with vinyl carbonate derivatives. By leveraging a sophisticated catalytic system comprising tris(dibenzylideneacetone)dipalladium and a specialized chiral Trost ligand, the process achieves remarkable enantioselectivity, with reported values reaching up to 97% ee. This level of stereochemical control is paramount for regulatory compliance in API manufacturing, where impurity profiles are strictly scrutinized. Furthermore, the reaction operates under mild conditions, typically at room temperature, which drastically reduces energy consumption compared to cryogenic or high-temperature alternatives. This combination of high efficiency, operational simplicity, and superior product quality positions this technology as a cornerstone for modern nucleoside production.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional strategies for constructing chiral acyclic nucleosides have long been hindered by their inherent inefficiency and reliance on scarce resources. The first conventional pathway involves the meticulous design and synthesis of a chiral acyclic side chain, often derived from natural amino acids or sugars, followed by coupling to the nucleobase via nucleophilic substitution or Mitsunobu reactions. This approach is not only step-intensive but also suffers from poor atom economy, as the chiral source is often used in stoichiometric amounts and partially discarded during the linking process. The second pathway attempts to build the base onto a pre-functionalized chiral side chain, yet this too demands complex protection-deprotection sequences and expensive starting materials. Consequently, these legacy methods result in prolonged lead times, elevated waste generation, and a fragile supply chain vulnerable to fluctuations in the availability of natural chiral feedstocks.
The Novel Approach
In stark contrast, the novel asymmetric allylation strategy described in the patent offers a direct, convergent route that fundamentally simplifies the molecular architecture assembly. By reacting readily available substituted purines or pyrimidines directly with vinyl carbonate, the method installs the chiral hydroxy-allyl side chain in a single catalytic step. This eliminates the need for pre-formed chiral side chains entirely, replacing them with inexpensive, achiral commodity chemicals. The reaction is driven by a highly active palladium catalyst system that facilitates the formation of the carbon-nitrogen bond with simultaneous creation of the chiral center. This streamlining of the synthetic route not only accelerates the time-to-market for new drug candidates but also provides a robust platform for scaling up production without the bottlenecks associated with multi-step chiral pool syntheses.
Mechanistic Insights into Pd-Catalyzed Asymmetric Allylation
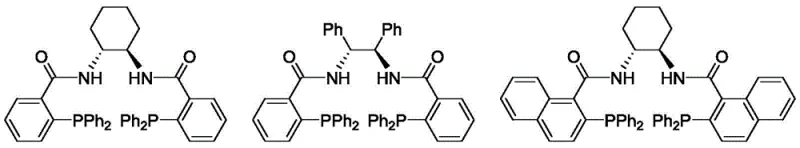
The success of this transformation hinges on the intricate interplay between the palladium catalyst and the chiral ligand environment. The mechanism initiates with the oxidative addition of the palladium(0) species to the vinyl carbonate, generating a cationic pi-allyl palladium complex. This intermediate is electrophilic and susceptible to nucleophilic attack by the nitrogen atoms of the purine or pyrimidine base. The critical element governing the stereochemical outcome is the chiral Trost ligand, specifically the naphthyl-based variant L8. This ligand coordinates to the palladium center, creating a rigid, chiral pocket that differentiates the two enantiotopic faces of the allyl moiety. As the nucleobase approaches, steric repulsion from the bulky binaphthyl backbone of the ligand forces the attack to occur from a specific trajectory, thereby locking in the desired (R) or (S) configuration with high fidelity.
Beyond stereocontrol, the choice of ligand L8 also plays a pivotal role in suppressing regioisomeric impurities. Purine bases, for instance, possess multiple nucleophilic nitrogen sites (N7 vs N9), and uncontrolled reactions often yield mixtures that are difficult to separate. The specific electronic and steric properties of the L8-palladium complex favor attack at the N9 position of the purine ring, ensuring high regioselectivity alongside enantioselectivity. This dual control mechanism is essential for producing pharmaceutical-grade intermediates, as it minimizes the burden on downstream purification processes. The ability to tune the reaction outcome simply by switching between the (1R,2R) and (1S,2S) enantiomers of the ligand further enhances the versatility of this platform, allowing access to both enantiomers of the target nucleoside from the same set of achiral starting materials.
How to Synthesize Chiral Acyclic Nucleosides Efficiently
Implementing this advanced synthesis protocol requires careful attention to reaction parameters to maximize yield and optical purity. The process is designed to be operationally simple, avoiding the need for specialized high-pressure equipment or extreme temperatures. The standard procedure involves combining the nucleobase substrate and vinyl carbonate in a polar aprotic solvent, with acetonitrile identified as the optimal medium for balancing solubility and reaction rate. The addition of the catalyst and ligand under an inert atmosphere initiates the transformation, which typically reaches completion within 12 hours at ambient temperature. Following the reaction, a straightforward aqueous workup and chromatographic purification yield the target compound. For detailed operational parameters and specific molar ratios optimized for different substrates, please refer to the standardized synthesis guide below.
- Combine substituted purine or pyrimidine substrate with vinyl carbonate in an organic solvent such as acetonitrile under an inert nitrogen atmosphere.
- Add tris(dibenzylideneacetone)dipalladium catalyst and chiral Trost ligand L8, then stir the mixture at room temperature for approximately 12 hours.
- Quench the reaction, extract with dichloromethane and water, dry the organic phase, and purify the crude product via column chromatography to obtain the target chiral nucleoside.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this asymmetric allylation technology offers profound advantages for cost management and supply chain resilience. The shift from stoichiometric chiral reagents to a catalytic system represents a fundamental change in the cost structure of nucleoside manufacturing. By utilizing low loadings of palladium catalyst and ligand, the process significantly reduces the consumption of high-value chiral materials. Furthermore, the use of vinyl carbonate as a versatile allylating agent replaces more hazardous or expensive alkylating agents, contributing to a safer and more cost-effective operational profile. These factors combine to lower the overall cost of goods sold (COGS), making the final API more competitive in the global marketplace.
- Cost Reduction in Manufacturing: The elimination of multi-step chiral side chain synthesis removes several unit operations from the production workflow, including isolation, purification, and quality control of intermediates. This consolidation of steps leads to substantial savings in labor, solvent usage, and facility time. Additionally, the high atom economy of the allylation reaction ensures that a greater proportion of the raw material mass is incorporated into the final product, minimizing waste disposal costs. The ability to operate at room temperature further reduces utility expenses associated with heating or cooling large-scale reactors, providing a cumulative financial benefit that scales with production volume.
- Enhanced Supply Chain Reliability: Dependence on natural chiral pools often exposes manufacturers to supply volatility driven by agricultural yields or geopolitical factors. In contrast, the raw materials for this process—substituted purines, pyrimidines, and vinyl carbonates—are commodity chemicals produced on a massive industrial scale with stable pricing and consistent availability. This decoupling from biological feedstocks ensures a more predictable and secure supply chain, reducing the risk of production delays due to raw material shortages. The robustness of the reaction across a wide range of substrates also allows for flexible sourcing of different nucleobase variants without requalifying the entire process.
- Scalability and Environmental Compliance: The mild reaction conditions and simplified workup procedure make this technology highly amenable to scale-up from kilogram to multi-ton production. The absence of harsh reagents or extreme conditions lowers the safety risks associated with large-scale operations, facilitating easier regulatory approval for manufacturing sites. Moreover, the reduction in solvent consumption and waste generation aligns with increasingly stringent environmental regulations and corporate sustainability goals. The high selectivity of the reaction minimizes the formation of difficult-to-remove impurities, reducing the need for extensive recrystallization or chromatography, which are often the most waste-intensive steps in pharmaceutical manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this patented synthesis method. These insights are derived directly from the experimental data and optimization studies presented in the patent documentation, providing a clear understanding of the technology's capabilities and limitations. Understanding these details is crucial for R&D teams evaluating the feasibility of integrating this route into their existing development pipelines.
Q: What represents the primary advantage of this asymmetric allylation method over traditional chiral pool synthesis?
A: Unlike traditional methods that rely on expensive, pre-existing chiral side chains and multi-step coupling reactions, this patented approach utilizes readily available achiral raw materials. It constructs the chiral center directly during the bond-forming event via catalytic asymmetric allylation, significantly reducing step count and raw material costs while achieving high enantiomeric excess (up to 97% ee).
Q: How does the choice of Trost ligand L8 impact the stereoselectivity of the reaction?
A: The naphthyl Trost ligand L8 provides a specific chiral environment around the palladium center that dictates the facial selectivity of the allylation. Its bulky binaphthyl backbone creates significant steric differentiation between the prochiral faces of the pi-allyl palladium intermediate, ensuring that the nucleophilic attack by the purine or pyrimidine base occurs with high regio- and enantioselectivity, minimizing the formation of unwanted isomers.
Q: Is the deprotection of protected amino groups compatible with the final product stability?
A: Yes, the patent demonstrates that protecting groups such as bis-tert-butoxycarbonyl on the amino substituents can be cleanly removed using sodium tert-butoxide in methanol. This deprotection step proceeds under mild conditions without compromising the integrity of the newly formed chiral allylic bond or the nucleobase, yielding the free amine products with retained high optical purity.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral Acyclic Nucleosides Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient chiral synthesis in the modern pharmaceutical landscape. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative laboratory discoveries are seamlessly translated into reliable industrial reality. Our state-of-the-art facilities are equipped to handle sensitive palladium-catalyzed reactions with rigorous safety protocols, while our stringent purity specifications and rigorous QC labs guarantee that every batch of chiral acyclic nucleosides meets the highest international standards. We are committed to supporting your drug development journey with scalable, cost-effective solutions that accelerate your time to market.
We invite you to collaborate with us to leverage this cutting-edge asymmetric allylation technology for your next-generation antiviral programs. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific project requirements, demonstrating how this route can optimize your manufacturing economics. Please contact our technical procurement team today to request specific COA data for our reference standards and to discuss comprehensive route feasibility assessments for your target molecules. Let us be your partner in transforming complex chemical challenges into commercial successes.